RESEARCH研究テーマ
- AI創薬・インシリコ創薬
-
- 医薬品候補化合物の構造生成
- 創薬ターゲットの同定
- 医薬品候補化合物の薬効や副作用の予測
- ドラッグリポジショニング、エコファーマ
- 薬物・標的タンパク質間相互作用の予測
- シナジー創薬学
- 薬物の組み合わせ効果の予測
- 精密医療
-
- AI医療
- 患者層別化
- 疾患サブタイプ分類
- 病態バイオマーカーの同定
- シングルセル・オミックスデータの解析
- 再生医療
-
- 再生医療におけるAI手法の開発
- ダイレクトリプログラミングによる再生医療
- 細胞直接変換を誘導する転写因子の予測
- 細胞直接変換を誘導する低分子化合物の予測
- 低分子化合物によるダイレクトリプログラミング
- ヘルスケア
-
- 一般化合物の毒性・副作用の予測
- 健康食品・化粧品のメカニズムの解明
- 食品成分・化粧品成分の効能の予測
- 漢方薬のメカニズムの解明
- 漢方薬のリポジショニング
- システム生物学
-
- 教師付き学習による分子ネットワークの推定
- 遺伝子制御ネットワークの予測
- 代謝パスウェイの予測
- タンパク質間相互作用ネットワークの予測
- タンパク質・リガンド相互作用ネットワークの予測
- バイオインフォマティクス
-
- ミッシング酵素遺伝子の同定
- 多階層オミックスデータの融合
- 遺伝子発現データの統計解析
- オーソログ遺伝子クラスターの自動構築
- 糖鎖の分類やモチーフ抽出
- ケモインフォマティクス
-
- 酵素反応の予測
- 酵素番号の自動割り当て
- ヴァーチャルスクリーニング
- 化合物の分類
- 化合物や反応の類似性の設計
- 統計学・機械学習
-
- カーネル法
- グラフ推定
- スパース統計モデル
- 感度分析
- 関数データ解析
研究内容の具体的な紹介いくつかの研究内容について、以下で紹介します。[研究紹介動画]
- 医薬品候補化合物の構造生成AI
-
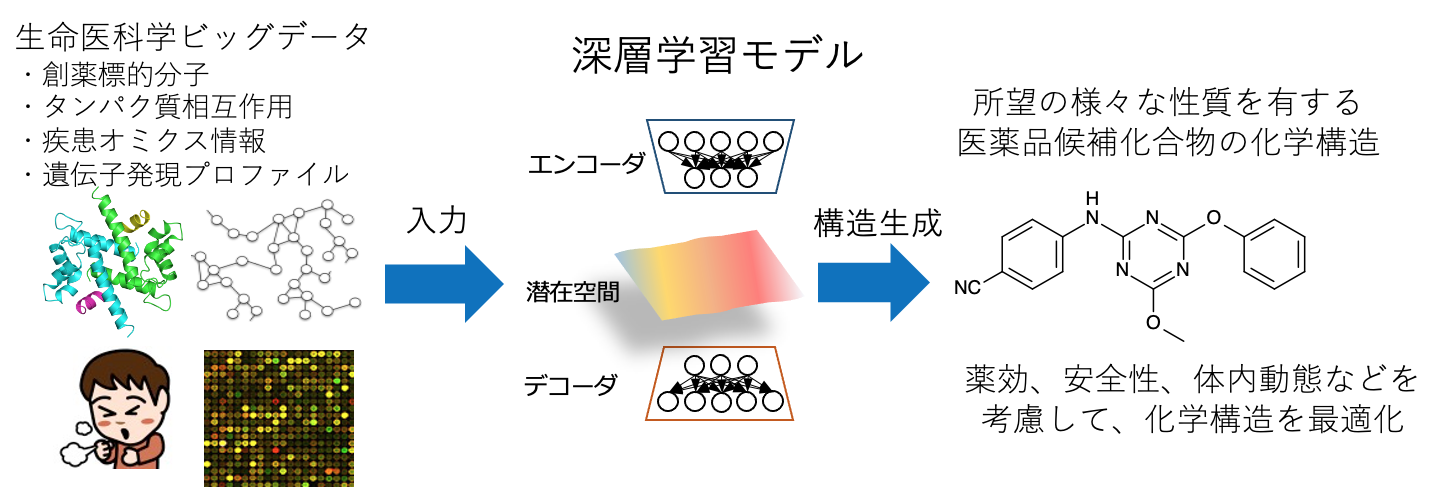
本研究では、化学分野や生命医科学分野のビッグデータを解析し、医薬品候補となる低分子化合物や中分子化合物(ペプチドなど)の新規構造を生成するAIの開発を行っています。特に、所望の生物活性やオミクス情報(創薬標的遺伝子ノックダウンのトランスクリプトームなど)を入力とし、その性質を満たす化合物を自動で出力する深層学習モデルが特長です。構造生成の先行研究はありますが、化学構造の変換が不確実であり、生成する構造の多様性や、生物活性や毒性などが考慮されていません。生物活性・毒性情報やオミクス情報などの複数の性質を満たす化学構造を発生することで、自律的に分子設計を行います。新薬の開発期間短縮や開発費用の削減により、薬価の高騰の抑制や国家の医療費の削減にも貢献することが期待できます。
遺伝子発現プロファイルなどのオミクス情報とケミカル情報を融合し、変分オートエンコーダ(VAE)や類似度検索などを組み合わせて構造発生AIを開発しました。例えば、がんの創薬標的分子 10 種類に対して、それらのターゲット遺伝子摂動応答遺伝子発現プロファイルからのリガンドの構造発生を試み、validity, uniqueness, novelty などの指標で既往手法よりも優れた値を示しました。他の手法と比較して、よりリガンドらしい構造式を発生させることができました(Kaitoh et al, J Chem Inf Model, 2021)。
生成構造の多様性を確保するため、ビシクロ[1.1.1]ペンタンやキュバンなどの未踏骨格や、ホウ素やケイ素などの未踏原子を部分構造として網羅的に発生させる構造生成器を開発しました。部分構造を固定させる他の手法と比較して、提案手法は網羅的に構造式を発生させることに成功しました(Kaitoh et al, J Chem Inf Model, 2022)。
Transformerを敵対的生成ネットワーク(GAN)に導入し、所望の性質を満たす医薬品候補化合物の化学構造を強化学習ベースで発生させる構造生成器を開発しました(Li et al, IJCAI2022, 2022)。validity, uniqueness, novelty, diversity, 水溶性、医薬品らしさ、合成可能性などの視点で、提案手法の有用性を示しました。
- 複数の薬剤の組み合わせ効果(薬剤シナジー)を明らかにするシナジー創薬学
-
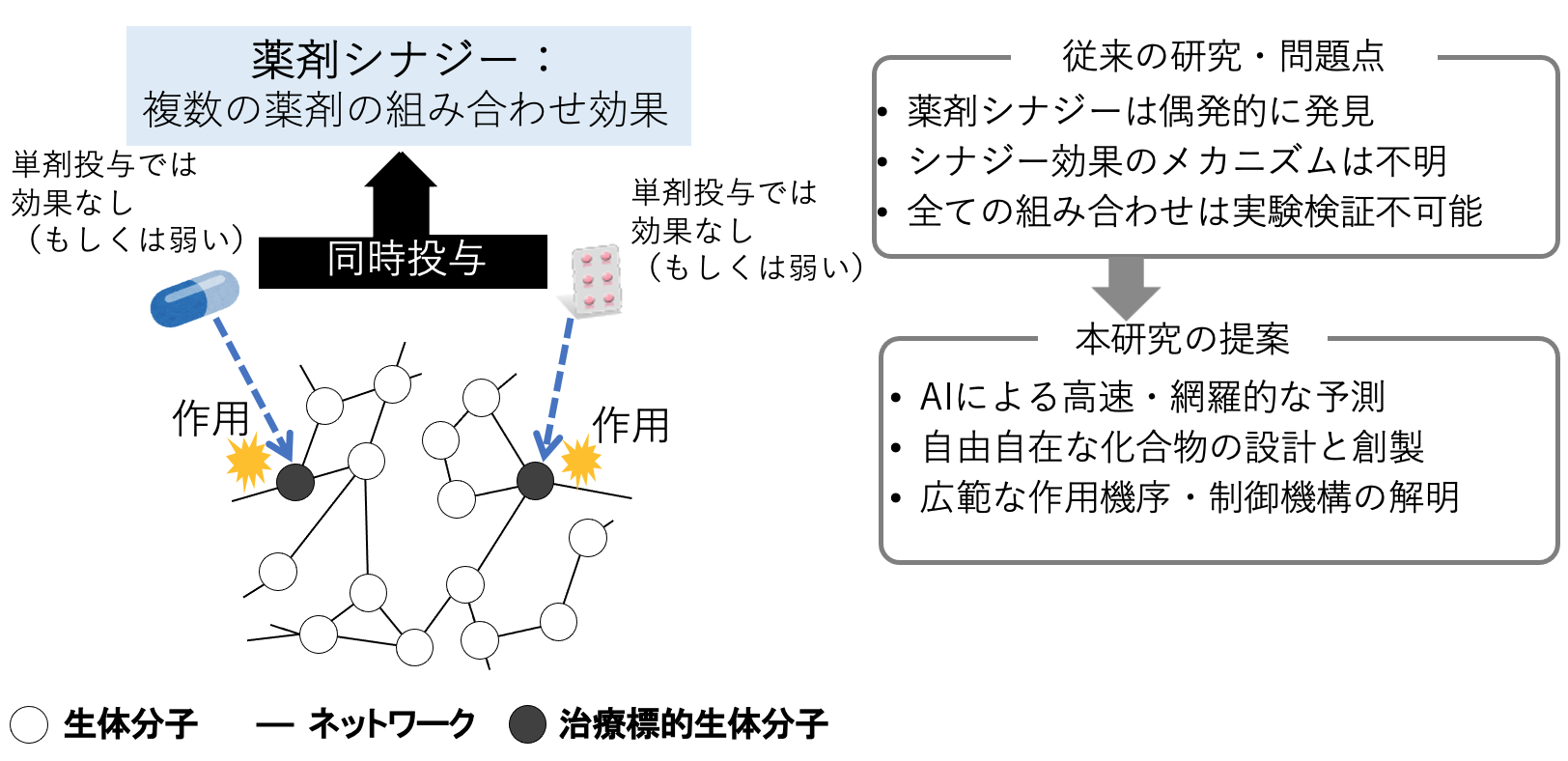
複数の薬剤の組み合わせによる相乗効果(薬剤シナジー)を活用した化学療法が、がんや神経変性疾患など多因子疾患に対する有効な治療法として注目されています。治療効果の増強だけでなく、個々の薬剤の使用量を減らし、重篤な副作用の発現頻度を低下させるなどの利点があり、これまでの治療法を一新させる可能性があります。しかしながら、やみくもな薬剤の組み合わせは有害な副作用に繋がるため、最適な薬剤の組み合わせを同定する必要がありますが、極めて困難です。これまでに報告されてきた薬剤シナジーは、臨床研究で偶発的に発見されたものが多く、疾患特異的な薬剤シナジーの発現メカニズムはよく分かっていませんでした。薬剤シナジーは、薬剤群と生体分子群の相互作用によって生み出されると考えられますが、どの生体分子(治療標的分子)への作用の組み合わせが薬物シナジーに繋がるかは不明です。本研究では、化学・生命関連ビッグデータを有効利用し、薬剤シナジーを生み出す最適な薬物の組み合わせを予測するAI手法を開発します。
詳細は、以下の学術変革B「シナジー創薬学」の領域HPをご覧ください。
[学術変革B「シナジー創薬学」の領域HP]
- 機械学習と医薬ビッグデータ解析によるAI創薬
-
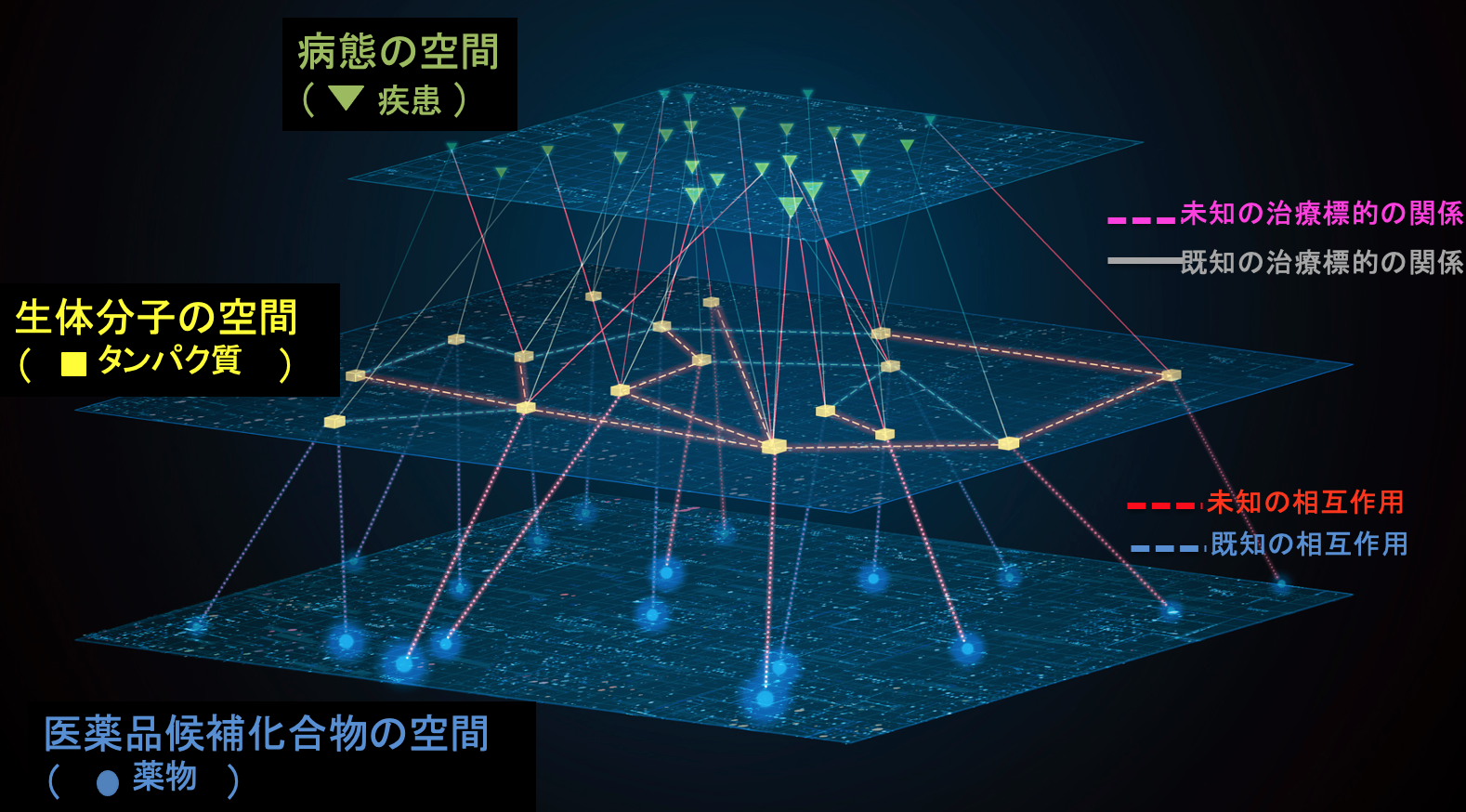
様々な疾患に対する多階層オミクスデータから疾患の共通性や特異性を見出し、治療標的分子や医薬品候補化合物を自動的に予測する機械学習の手法を開発しています。様々な疾患と分子間相互作用ネットワーク情報を紐付けすることによって、最良の治療標的分子や医薬品候補化合物を理論的に導出したり、医薬品候補化合物の化学構造を新しく設計する点が特色です。新薬の開発期間短縮や開発費用の削減により、薬価の高騰の抑制や国家の医療費の削減にも貢献することが期待できます。
医薬品開発において、治療標的分子(薬剤で制御することで疾患の治療に繋がる生体分子)を同定することは重要課題です。しかし、既存の病理学的知識から推測できる治療標的分子は限定されており、治療標的分子の枯渇が世界的な課題となっています。本研究では、既存の薬剤を新しい疾患に転用するドラッグリポジショニングの概念を治療標的分子へと拡張することで、既存の治療標的分子を新しい疾患に転用するターゲットリポジショニングの概念を提案しました(Namba et al, Bioinformatics, 2022)。提案手法は、治療標的分子の細胞応答を反映する遺伝子発現パターンと疾患特異的な遺伝子発現パターンの融合解析により、疾患横断的に治療標的分子を予測する機械学習手法を開発しました。遺伝子配列の変異情報に依存した従来手法では、疾患治療に向けて阻害すべき治療標的分子と活性化すべき治療標的分子を分けて予測することが困難でしたが、遺伝子発現情報を用いることで両者の識別を可能にしました。また、疾患間で類似する発症メカニズムを考慮することで、従来の手法と比較して治療標的分子を高い精度で予測することが可能となりました。提案手法は既存の治療標的分子だけでなく未開発の生体分子に対しても治療標的分子としての治療可能性を予測できるため、既存の治療標的分子の転用や新規の治療標的分子の創出による、医薬品開発の促進が期待されます。
詳細は、以下のプレスリリースのページをご覧ください。
[press release] [journal site]
- ダイレクトリプログラミングを誘導する転写因子の予測と再生医療への応用
-
再生医療のため、すでに分化した細胞を別の種類の細胞にiPS細胞を介さずに直接転換するダイレクトリプログラミングが注目されています。しかしながら、ダイレクトリプログラミングを誘導する転写因子を実験的に同定するのは、時間や実験コストの面から非常に困難なため、情報科学的な予測手法が切望されていました。
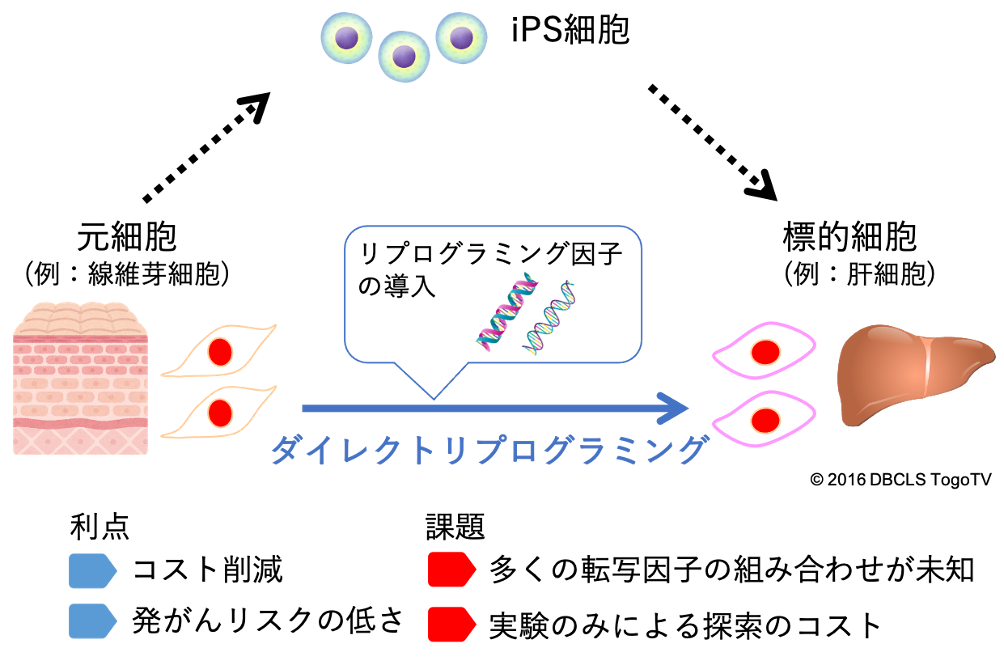
われわれは、トランスクリプトーム情報(ヒト細胞の遺伝子発現プロファイルなど)、エピゲノム情報(ChIP-seqによる転写因子DNA結合など)、ゲノム情報(エンハンサーなど)、インターラクトーム情報(遺伝子制御ネットワークなど)の多階層オミックスデータをトランスオミクス解析し、ダイレクトリプログラミングを誘導する転写因子を高精度に予測する情報技術を開発しました(Eguchi et al, Bioinformatics, 2022)。細胞変換に関与するエピジェネティックな修飾の変化を介して転写を活性化するパイオニア因子に着目し、クロマチン構造から標的の細胞種それぞれのパイオニア因子を推定しました。次いで、様々なマルチオミクス情報や細胞間の系統関係情報を統合し、パイオニア因子を考慮した上でダイレクトリプログラミングを誘導する転写因子を予測する世界初のコンセプトを提唱しました。
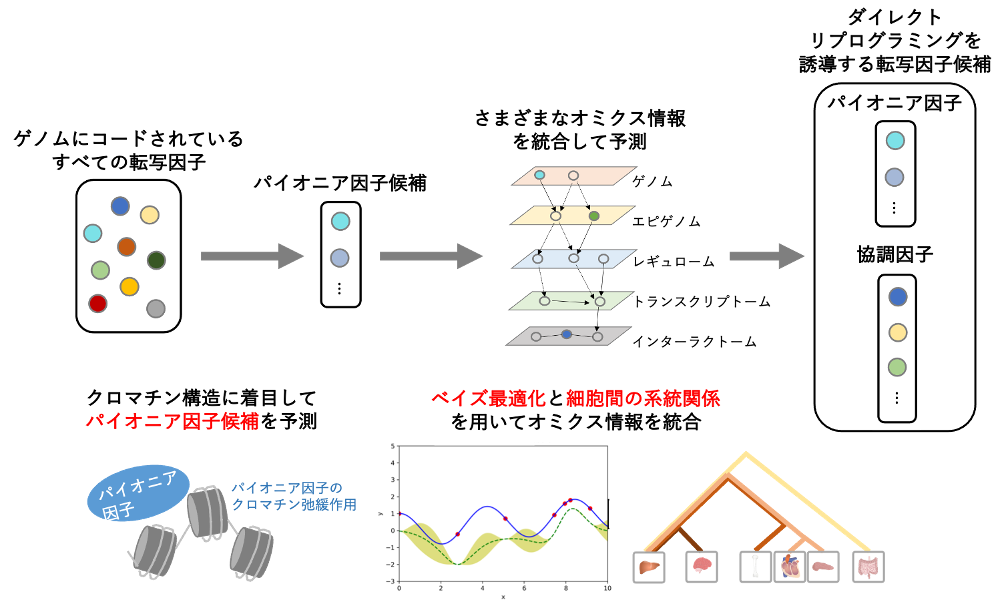
今回の提案手法では、皮膚由来の線維芽細胞を、さまざまな組織(肝臓、骨、脳、心臓、膵臓、腸)由来の細胞へと、それぞれ直接変換させる転写因子を予測しました。さらに、細胞変換メカニズムを考慮し、元細胞や標的細胞に関わる生体分子のマルチオミクスデータや細胞間の系統関係情報を、ベイズ最適化など機械学習アルゴリズムで統合することにより、ダイレクトリプログラミングを誘導する転写因子を高精度に予測することにも成功しました。
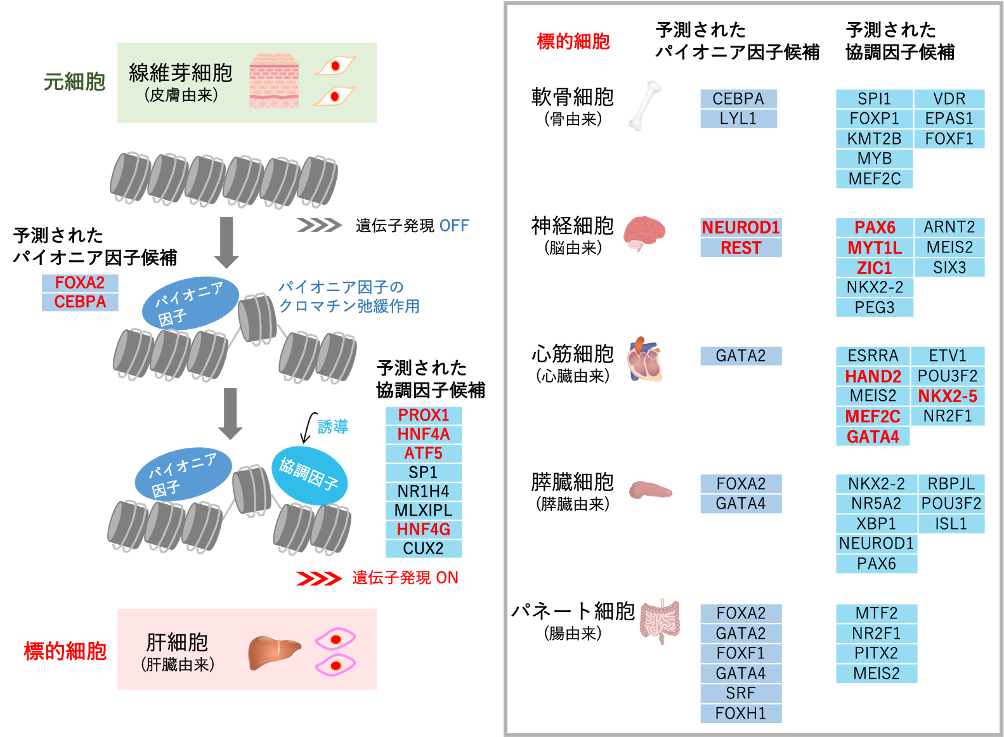
本提案手法により、ダイレクトリプログラミングを誘導する転写因子が未知の細胞変換に対しても、転写因子を新たに予測することができるようになり、今後の再生医療分野における細胞作製法の開発を促進することが期待されます。
詳細は、以下のプレスリリースのページをご覧ください。
[press release] [journal site]
- 低分子化合物でのダイレクトリプログラミングによる再生医療
-
再生医療のため、すでに分化した細胞を別の種類の細胞にiPS細胞を介さずに直接転換するダイレクトリプログラミングが注目されています。通常は外来遺伝子を導入する方法がとられていますが、ウィルスに起因する発がんリスクが問題となっていました。
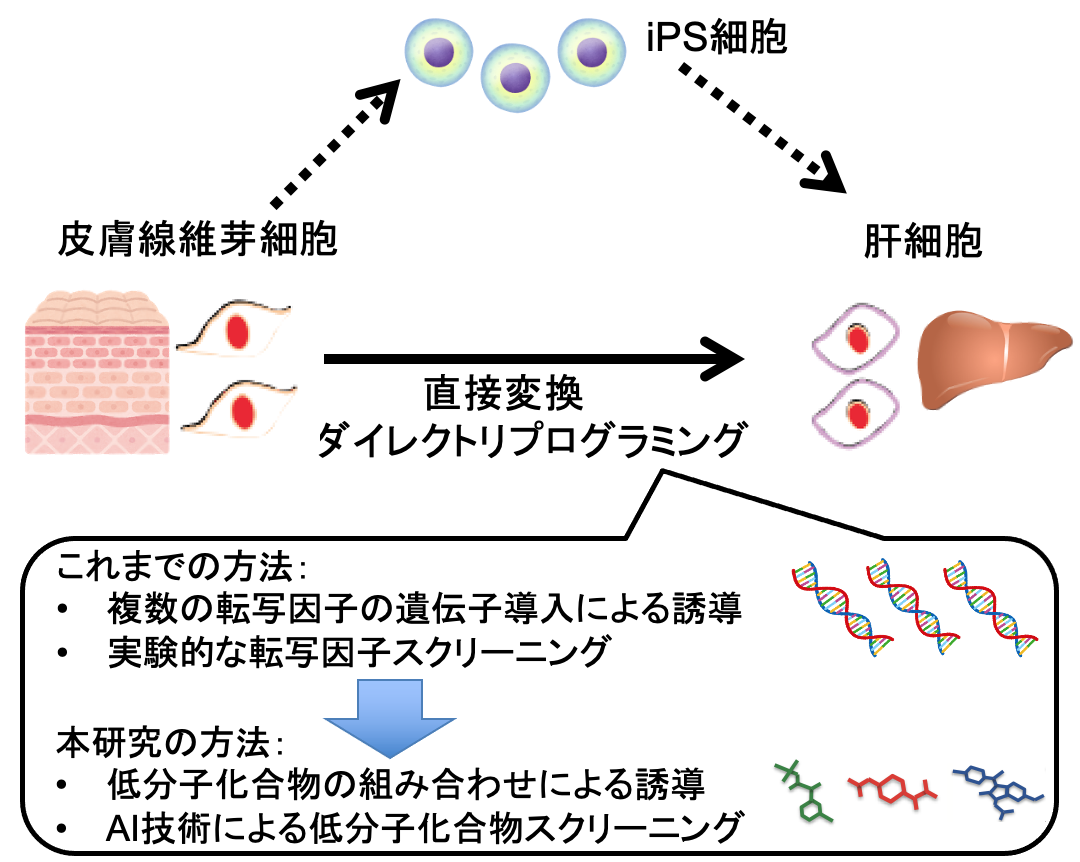
当研究室では、低分子化合物によるダイレクトリプログラミングのための機械学習の手法を開発しています。すでに分化した細胞を別の種類の細胞へと直接変換するダイレクトリプログラミングを誘導する低分子化合物を予測する手法を開発しました(Nakamura et al, Bioinformatics, 2022)。通常の細胞の直接変換は、ウイルスを用いて必要な遺伝子を元細胞に導入するため、ウイルスに起因する発がんリスクなどの問題がありました。そこで遺伝子導入の代わりに低分子化合物の添加による細胞変換を誘導する技術が切望されていますが、実験的に低分子化合物を同定するのは時間や実験コストの面から極めて困難です。そこで、細胞の直接変換を誘導する低分子化合物を予測する最適化アルゴリズムを開発しました。まず、細胞が変換する分子メカニズムに着目し、変換過程で重要な生物学的パスウェイを明らかにしました。次いで、その生物学的パスウェイを制御する低分子化合物の最適な組み合わせを探索することによって、細胞の直接変換を誘導する低分子化合物の新たな組み合わせを予測する手法の確立に成功しました。開発手法を用いて、皮膚線維芽細胞から神経細胞や心筋細胞への直接変換を誘導する低分子化合物の組み合わせを予測しまし、その有用性を示しました。本開発手法によって、細胞の直接変換を誘導する低分子化合物を簡便に予測することができるようになり、今後の再生医療分野における細胞作製法の安全性の向上を促進することが期待されます。
詳細は、以下のプレスリリースのページをご覧ください。
[press release] [journal site]
- トランスオミクス解析により、疾患の病態メカニズムをシングルセルレベルで解明
-
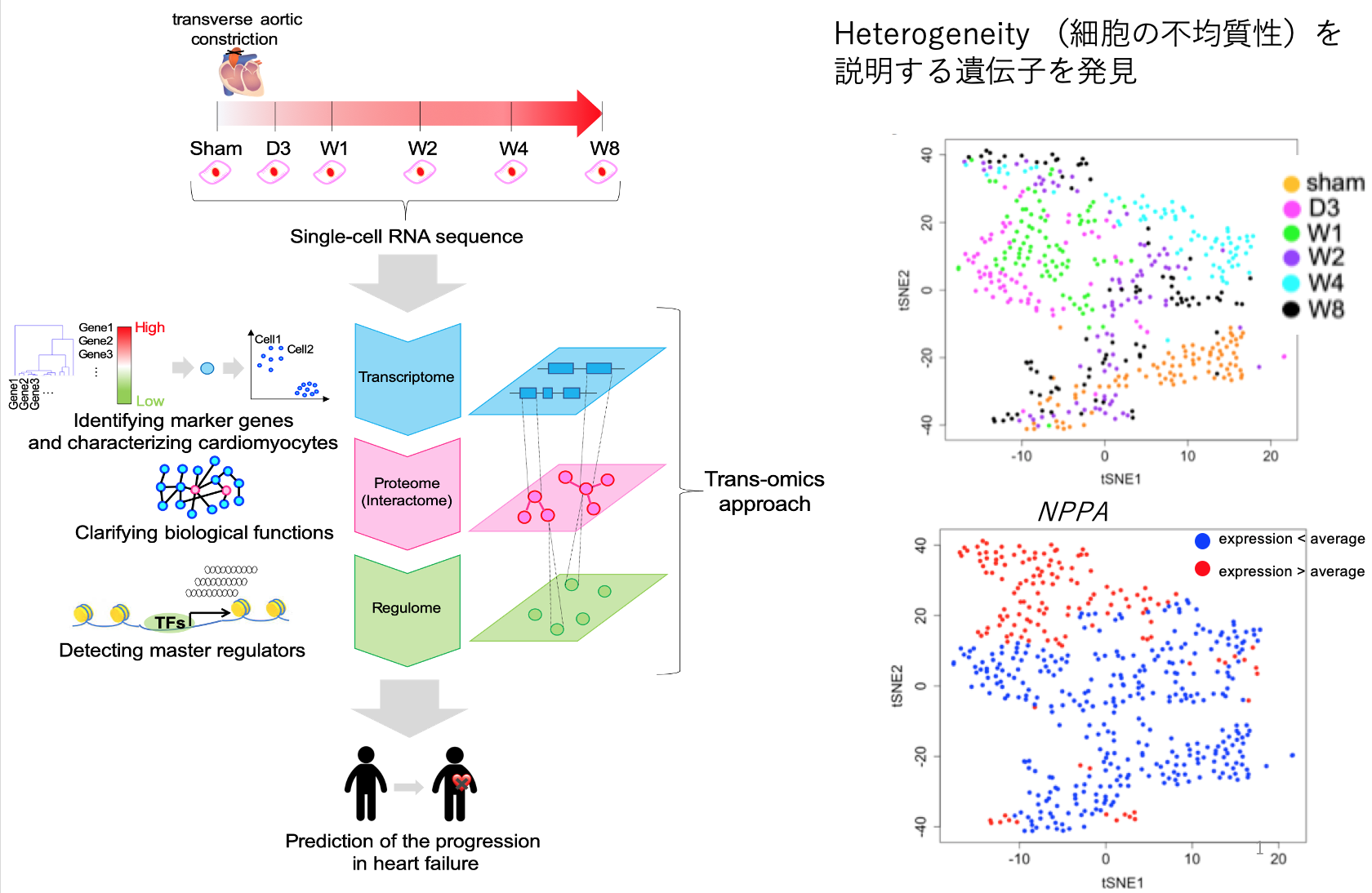
日本において心不全患者は2035年には132万人にも達すると推定されており、今後も増加することが問題視されています。心不全は複数の危険因子が存在し病態も多様であることから、発症や病態悪化のメカニズムは未だに分かっていません。そこで、本研究では心不全の分子病態メカニズムを一細胞レベルから明らかにすること目的としました(Hamano et al, Scientific Reports, 2021)。大動脈結索(TAC)により心不全を誘発したマウスの心筋から、Single-cell RNA-seqにより時系列の遺伝子発現データを取得し、トランスクリプトーム、インターラクトーム、レギュロームの多階層オミックスデータのトランスオミクス解析により、不均一性の大きい心筋細胞を特徴付けるマーカーを探索すると共に、細胞の特徴付けを行いました。細胞を特徴付けるマーカーとしてnatriuretic peptide A (Nppa)を同定し、Nppaの発現が高い細胞ではミトコンドリア機能が低下することが推察される特徴的な遺伝子発現パターンを示しました。心不全の分子病態メカニズムとして、心肥大発症初期におけるNppaの顕著な発現上昇とミトコンドリア機能不全を介した経路の一端を明らかにしました。
詳細は、以下の論文をご覧ください。
[journal site]
- 様々な疾患オミックスデータや分子ネットワークの融合解析で、創薬標的分子や治療薬を探索
-
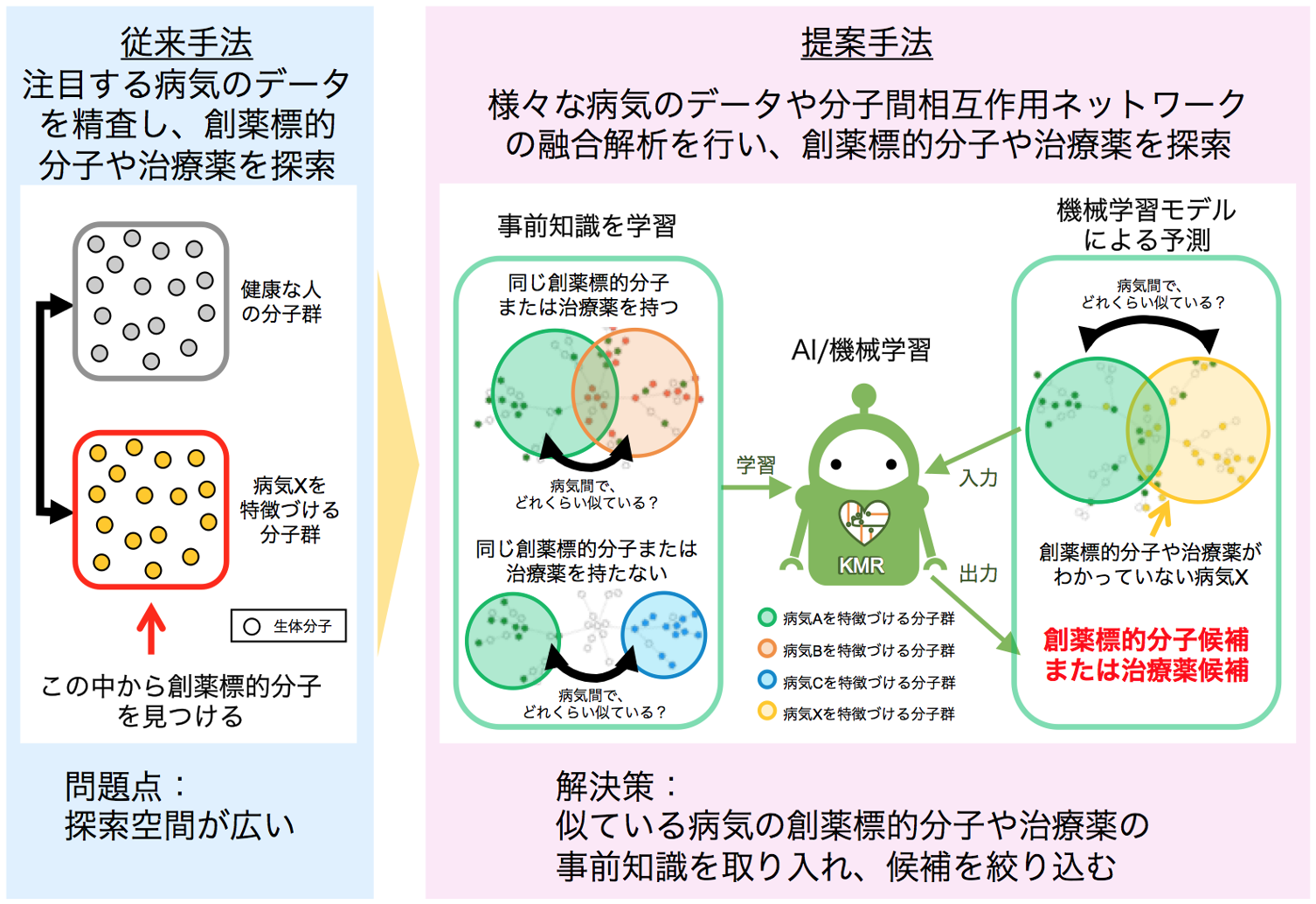
医薬品開発において、創薬標的分子やその制御化合物を見つけることは重要課題です。これまで、注目する病気のデータを精査し、創薬標的分子や治療薬の探索がされてきましたが、創薬標的の候補となる生体分子は非常に多く、探索空間が広いため、有効な創薬標的分子や治療薬を選ぶのが困難という問題がありました。本研究では、病気を特徴付けるさまざまな分子間相互作用ネットワークを比較することにより、分子間の機能的な連動性を考慮し、病気間の共通性や特異性を明らかにする方法を提案しました(Iida et al, Bioinformatics, 2020)。さらに、似ている病気の創薬標的分子や治療薬の事前知識を取り入れ、候補を絞り込むことにより創薬標的分子や治療薬を効率良く探索する機械学習アルゴリズム(AI基盤技術)を開発しました。これにより、従来法と比較して、創薬標的分子や治療薬を高い精度で探索することが可能となりました。開発手法は、創薬標的分子や治療薬の探索だけでなく、病気の分子メカニズムの解明、薬効予測などに活用できるため、医薬品開発に大きく貢献することが期待されます。
詳細は、以下のプレスリリースのページをご覧ください。
[press release] [journal site]
- パスウェイ制御による創薬
-
従来の創薬では一つの生体分子(タンパク質など)に注目し、それに特異的に作用する化合物を見つけていきます。しかしながら、その生体分子が関わる分子間相互作用ネットワークは考慮されておらず、単一の生体分子に特異的に作用する化合物が必ずしも期待する薬効に繋がるとは限りません。本研究では、従来の一つの生体分子の制御による創薬ではなく、多くの生体分子の機能モジュールであるパスウェイの制御による新しい創薬のコンセプトを、世界で初めて提唱しました(Iwata et al, J Med Chem, 2018)。
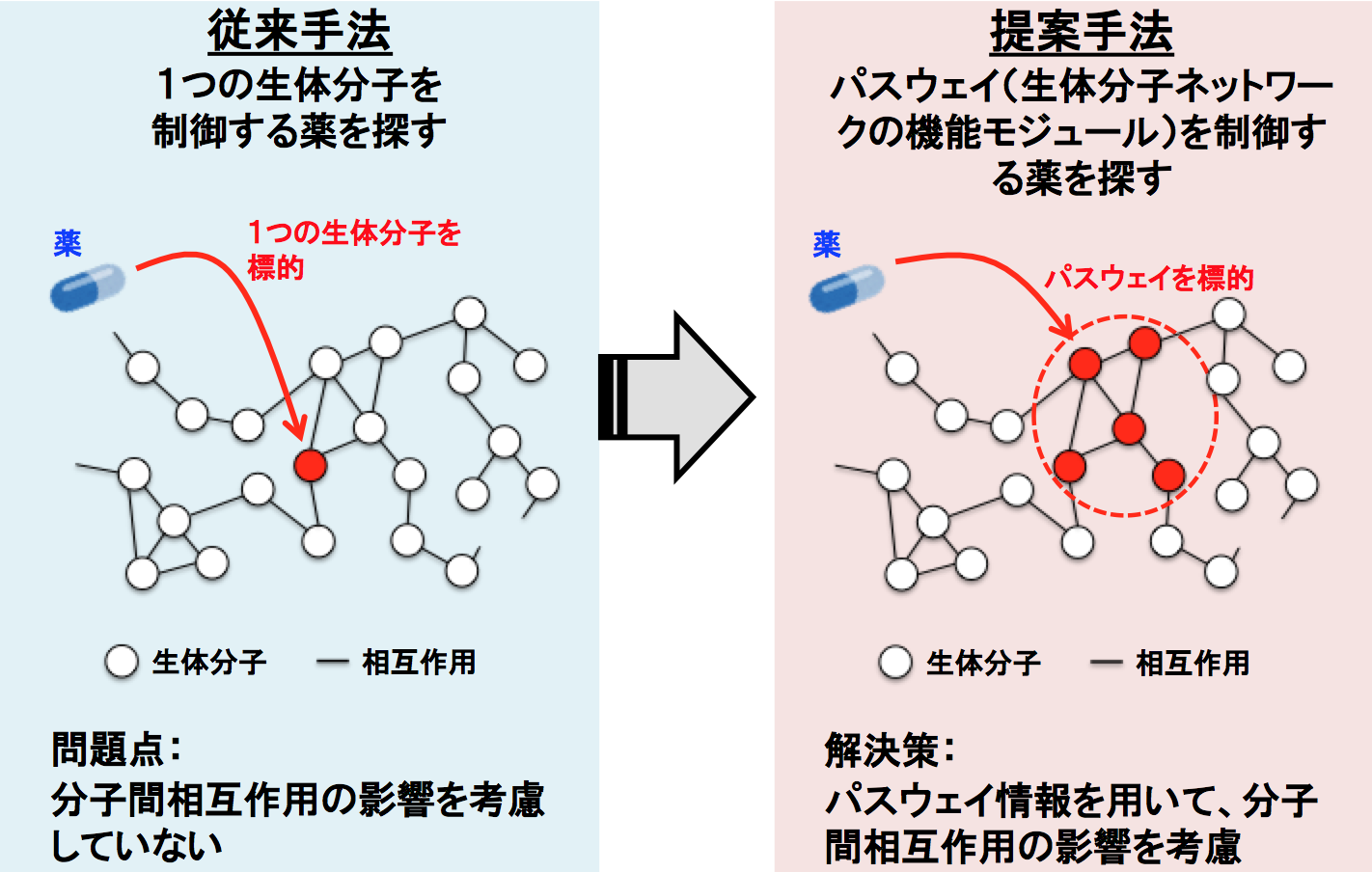
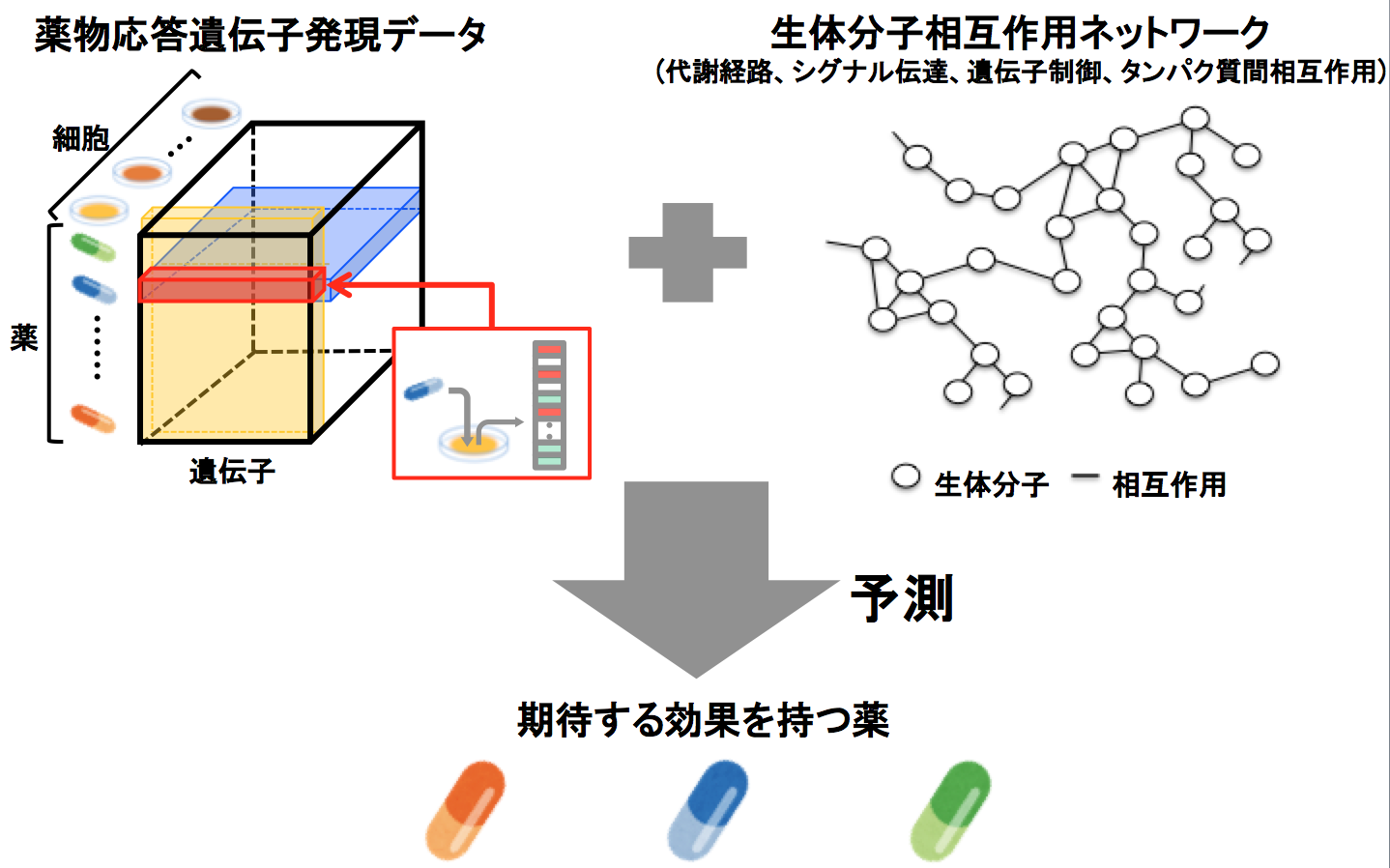
実際に、大規模な化合物応答遺伝子発現データと生体分子相互作用ネットワークの融合解析により、潜在的な抗がん作用を持つ化合物の同定に成功し、その有用性を示しました(Iwata et al, J Med Chem, 2018)。例えば、統合失調症治療薬であるペンルフリドールの抗がん作用(前立腺癌や直腸癌などに対する)を発見しました。
詳細は、以下のプレスリリースのページをご覧ください。
[press release] [journal site]
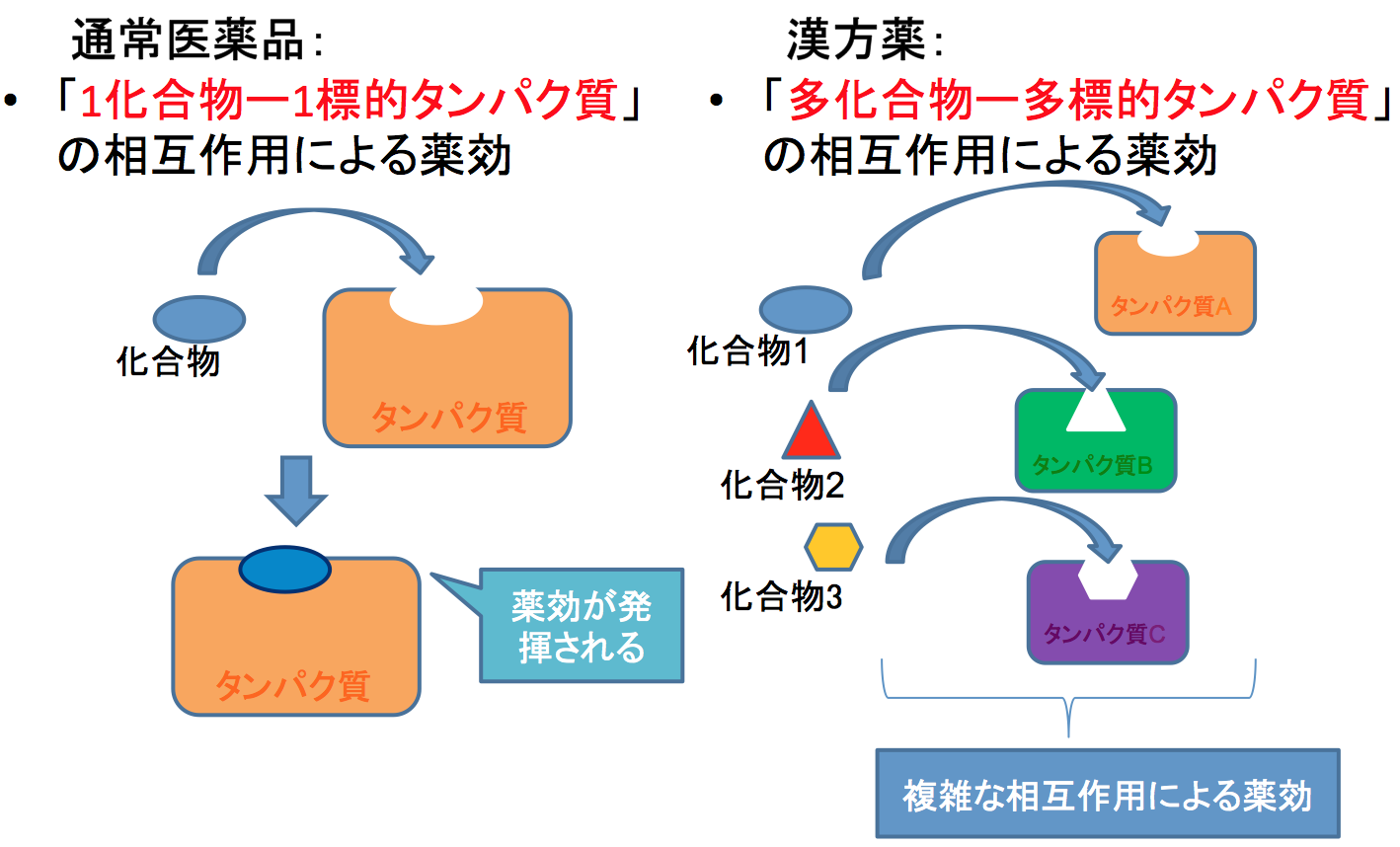
- 漢方薬リポジショニング
-
漢方薬の作用機序の解析や漢方薬の新しい適応可能疾患の予測を行うための情報技術を開発しています(Sawada et al, Sci Rep, 2018)。ドラッグリポジショニングの概念を漢方薬リポジショニングへ拡張するものです。
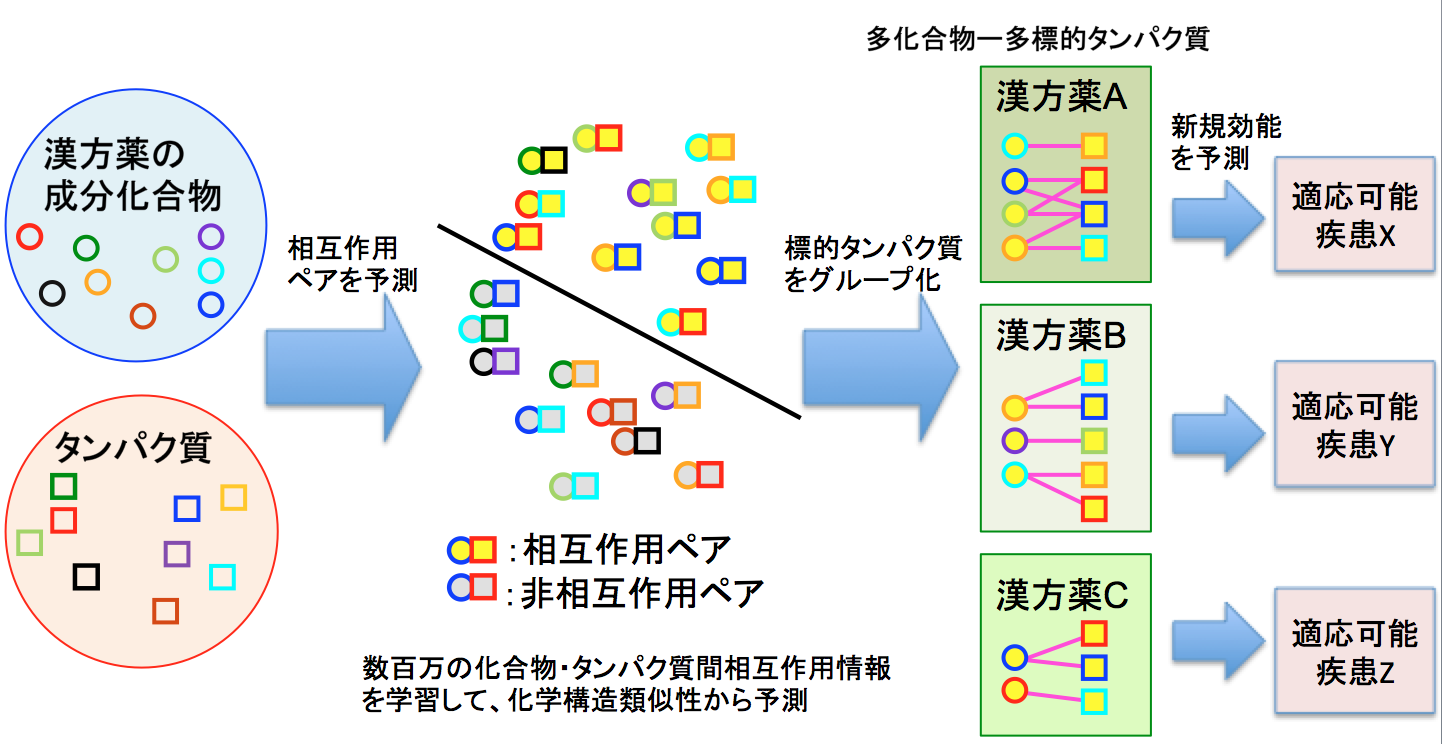
漢方薬の作用機序は複雑で多くの成分化合物の組み合わせが重要になりますが、成分化合物の標的分子群の分子機能やそれらのパスウェイの視点から漢方薬の作用機序を考察可能にしました(Sawada et al, Sci Rep, 2018)。例えば、肥満症の漢方薬である防已黄耆湯(ぼういおうぎとう)が、新たに糖尿病に対する効果がある可能性を示唆しました。このコンセプトは、食品や化粧品などへも展開可能です。
詳細は、以下のプレスリリースのページをご覧ください。
[press release][journal site]
- ドラッグリポジショニング
-
最近、承認される新薬の数が減っており、特にがんなどの難治性疾患の新薬開発が低迷しています。新薬を一個開発するのに数千億円の研究開発費と10年以上の歳月を要すると云われており、新薬の開発は極めて困難な道のりとなっています。そのような新薬開発の低迷を打開する創薬戦略として、既存薬の新しい効能を発見し別の疾患に対する治療薬として再開発するドラッグリポジショニング(Drug Repositioning)が注目を浴びています。既存薬はヒトでの安全性・体内動態・化合物製造法の情報が利用可能であるため、医薬品開発におけるいくつかの行程をスキップでき、開発期間が短いという特長があります。

ドラッグリポジショニングは既存薬の適応拡大の一種とも見なせます。例えば、シルデナフィル(バイアグラ)は狭心症の治療薬として当初開発されましたが、現在は勃起不全治療薬として使われています。ミノキシジル(ロゲイン、リアップ)は高血圧治療薬として当初開発されましたが、現在は増毛薬として使われています。しかしながら、このような過去の成功例は、偶然的な発見に大きく依存していました。
本研究では、近年の生命科学で大量に生み出されてきた薬物、遺伝子、タンパク質、疾患に関するビッグデータを有効利用し、データ科学的な根拠に基づくドラッグリポジショニングを目指しています。薬物に関するケミカル情報や副作用情報、薬物応答遺伝子発現情報,疾患に関するフェノタイプ情報やパスウェイ情報などの網羅的データを融合解析し,潜在的な薬物の効能を体系的に大規模予測するためのインシリコ手法の開発を行っています。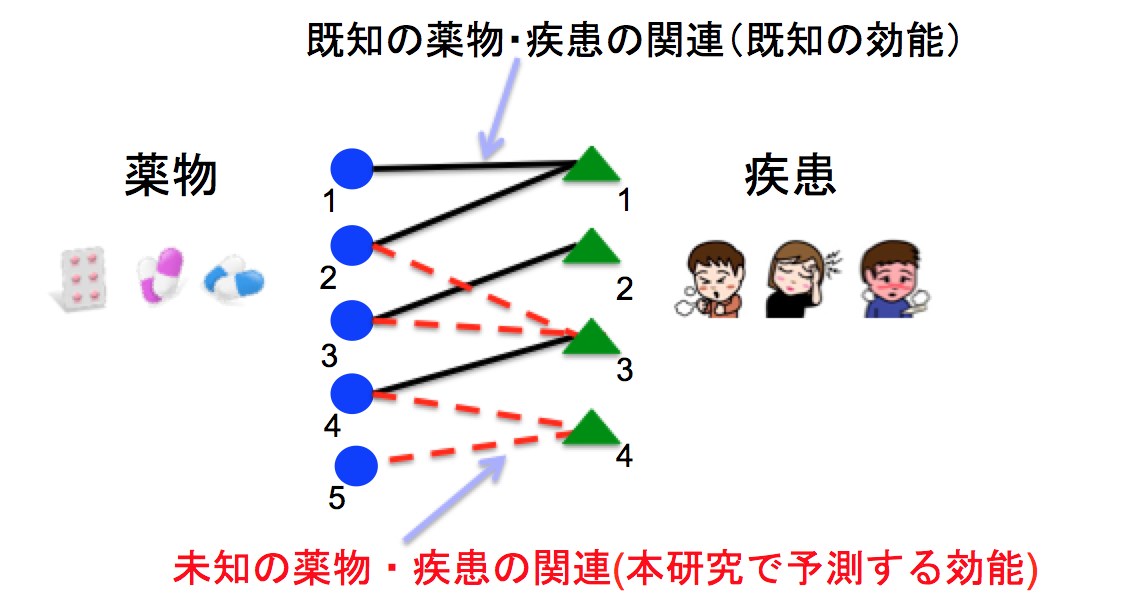
薬物の効能は薬物と疾患の関連として捉えることができます。ここでは,ドラッグリポジショニングの問題を多数の薬物群と多数の疾患群の間の関連ネットワークの予測問題に帰着させ、そのアルゴリズムを開発しています。提案手法を、日本や欧米で承認されている全ての薬物に適用し、潜在的な効能の大規模予測を行っています(Iwata et al, J Chem Inf Model, 2015)。また予測された新規効能の薬理作用機序まで解明するため、ケモゲノミクスや薬理ゲノミクスの手法を応用し、標的分子やオフターゲットタンパク質を介した既存薬の新規効能の大規模予測を行っています(Sawada et al, J Chem Inf Model, 2015)。
薬物の副作用データや疾患の分子関連データに適用したところ、2349個の薬物、858個の疾患に対して、117880個の新しい薬物・疾患関連ペアを予測することができました(Iwata et al, J Chem Inf Model, 2015)。以下の図は、様々な癌に対して予測された薬物・疾患の関連ネットワークの一部を表していて、本来癌の治療薬として開発されていない薬物が癌に効くと予測された例になります。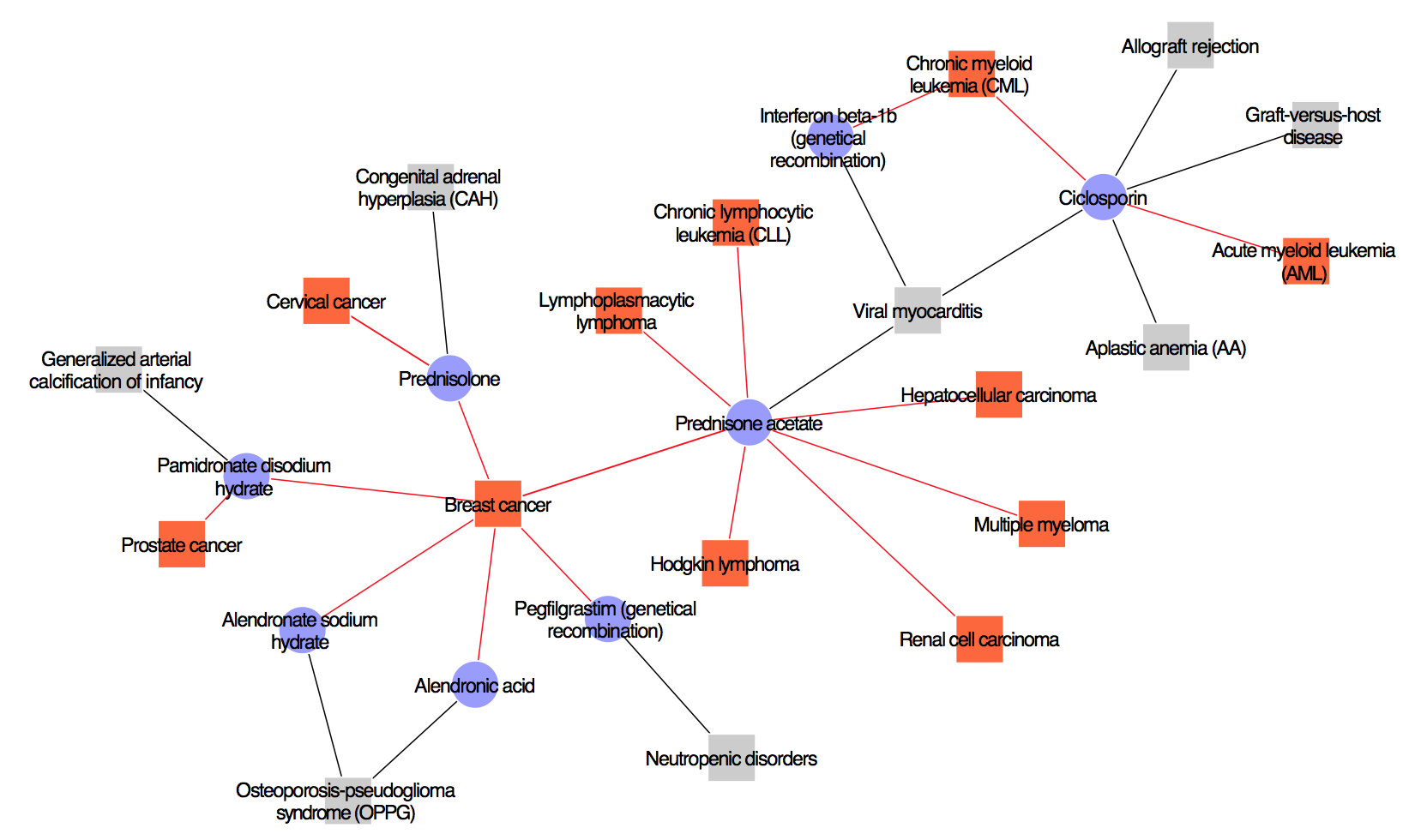
青色の丸いノードが薬物、オレンジ色の四角ノードが癌、灰色の四角ノードが癌以外の疾患、黒色のエッジが既知の薬物と疾患の関連(既知の効能)、赤色のエッジが新規に予測された薬物と疾患の関連(予測モデルの学習セットには入っていない効能)を表しています。例えば、アレンドロン酸(Alendronic acid)は骨粗鬆症(Osteoporosis−pseudoglioma syndrome)の薬ですが、乳がん(Breast cancer)に対する効能が予測されています。予測スコアが高い薬物・疾患ペアの多くは、近年の文献や臨床レポートで妥当性を確認することができました(上位40個中17個)。上の図では近年の文献等で確認がとれたものだけを示していますが、確認がとれなかったものは新しい医学的発見の可能性があり、予測された効能を臨床的に検証する価値が高いと考えられます(未公開データ)。実験系・臨床系の研究者、製薬会社との共同研究を募集しています。
- 薬と標的タンパク質間の相互作用ネットワークの予測
-
大半の医薬品(薬物)は標的とするタンパク質などの生体分子との相互作用を介して、薬効や副作用という形で人体に影響を及ぼします。そのため、薬物と標的タンパク質(オフターゲットを含む)の相互作用の網羅的同定は、医薬品の開発過程において最重要課題です。ポストゲノム研究では、遺伝子やタンパク質に関する大量のデータが得られるようになってきましたが、同時にハイスループットスクリーニングなどの技術の発展によって、膨大な数の化合物や薬物に関するケミカル情報や生理活性情報も蓄積されています。ヒトゲノム解析により全てのタンパク質をコードする数万のヒト遺伝子が明らかにされたにもかかわらず、実際に創薬のターゲットとして利用されているタンパク質はごく僅かです。また薬効や副作用に関与する相互作用のメカニズムもまだよく分かっていません。そのため、薬の候補化合物やタンパク質に関する様々なオミックスデータを有効活用し、薬物・標的タンパク質間相互作用ネットワークを大規模に予測するための情報技術の開発が期待されています。
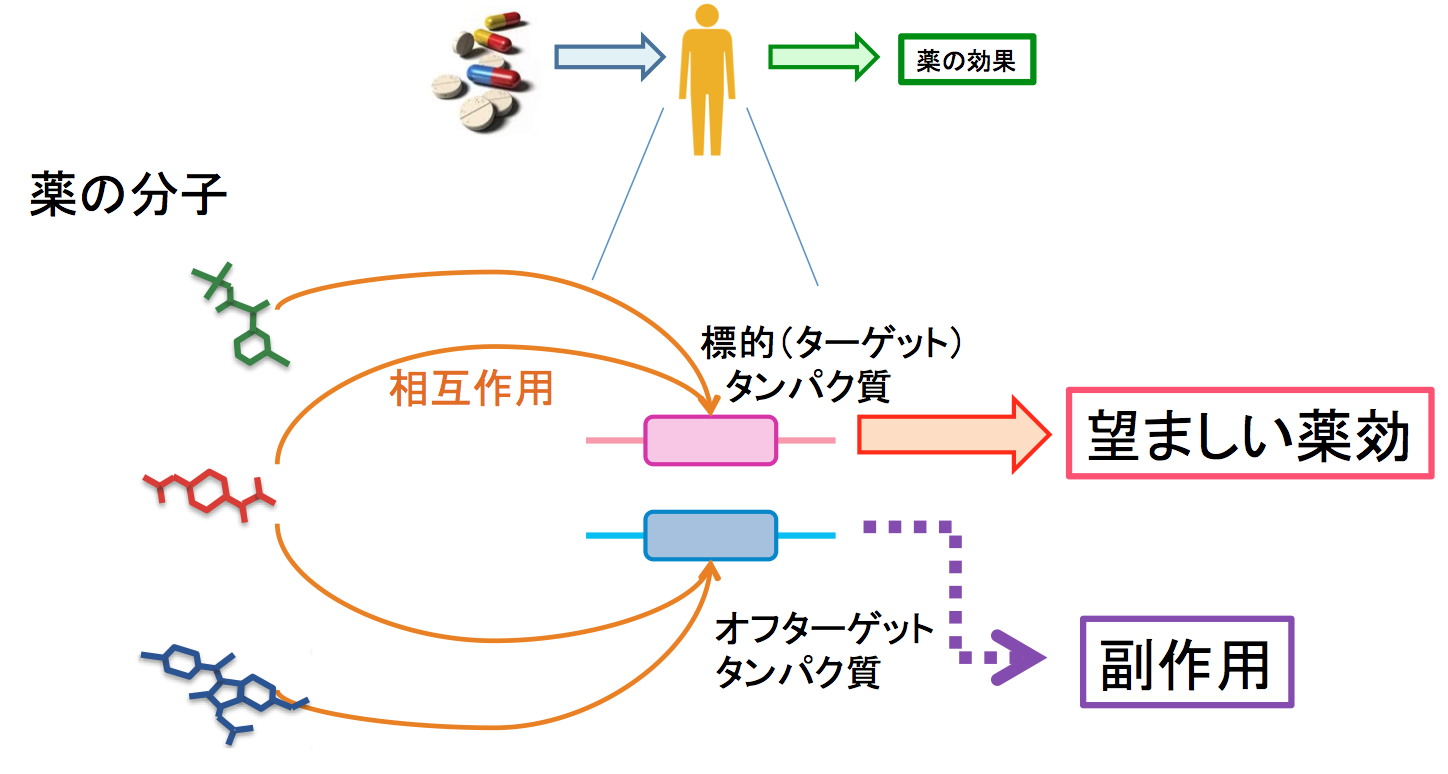
本研究では、薬の化学構造のケミカル空間、標的タンパク質のアミノ酸配列のゲノム空間、薬と標的タンパク質間の相互作用ネットワークの間の相関を解析しました。それをケミカルゲノミクスの観点からモデル化し、薬と標的タンパク質の間の未知の相互作用を大規模に予測する統計手法を開発しました(Yamanishi et al, Bioinformatics, 2008; Yamanishi, Adv Neural Inf Process Syst, 2009; Bleakley and Yamanishi, Bioinformatics, 2009)。下図のように、薬(または候補化合物)のセットとタンパク質のセットの関係を二部グラフで表現し、その潜在的な相互作用を推定する教師付き学習アルゴリズムを提案しました。
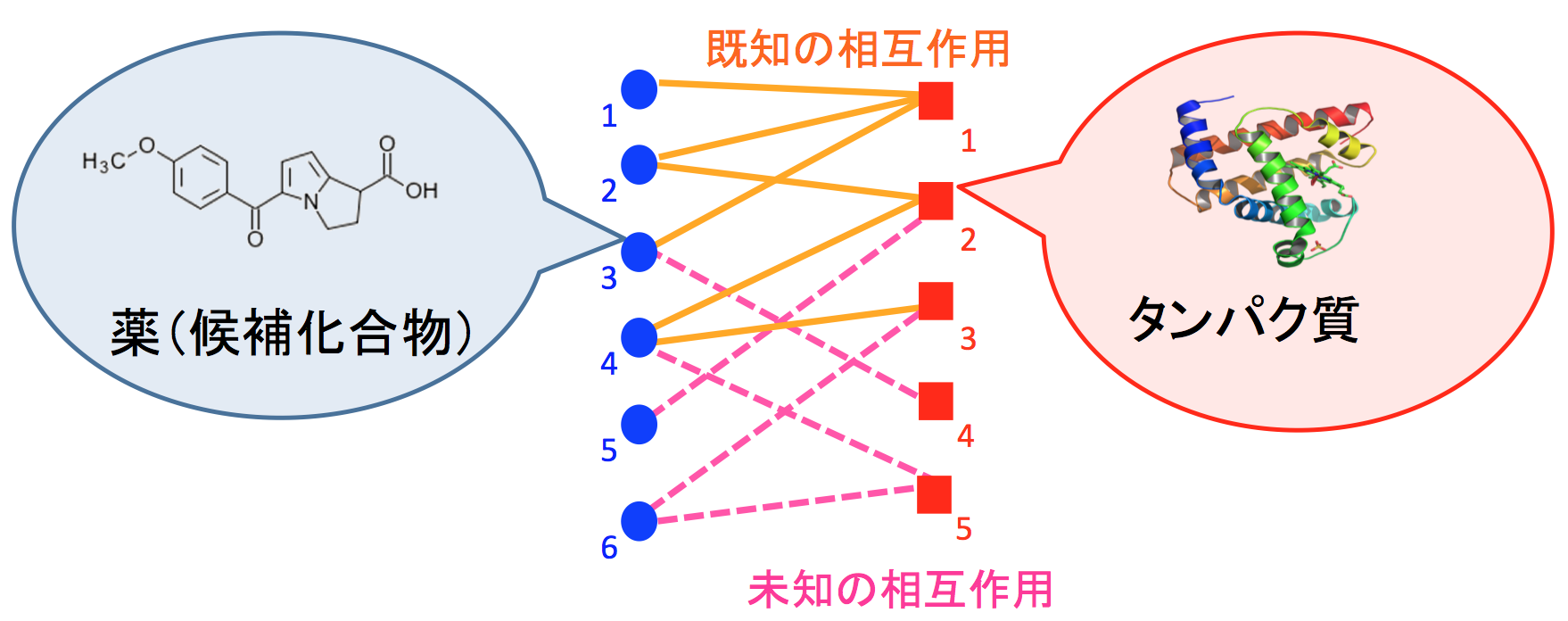
この手法は薬のケミカル情報とタンパク質のゲノム情報を融合させるケミカルゲノミクスの新しい手法で、現時点で世界最高峰の予測精度と網羅性を示しています。タンパク質の立体構造を必要とする従来のドッキングの手法とは異なり、興味のある薬や化合物の潜在的なターゲットをゲノムワイドに予測できる点が独創的です。
さらに、薬の化学構造、薬効・副作用情報、薬と標的タンパク質相互作用ネットワークの間の相関を検証し、薬効・副作用類似性が、化学構造類似性よりも、相互作用ネットワークに強く相関している事が分かりました。そして、薬効・副作用類似性とタンパク質の配列類似性に基づいて、薬と標的タンパク質の間の相互作用を予測する手法を開発しました(Yamanishi et al, Bioinformatics, 2010)。ケミカルゲノミクスの方法に比べ、化学構造からは想像もしないようなターゲットを予測できるのが特長です。
提案手法を、ヒトの酵素、イオンチャネル、G蛋白質共役型受容体、核内受容体に関するネットワークに適用し、クロスバリデーション実験で有効性を示しました。提案手法を、KEGGデータベースの全ての薬や化合物に適用し、薬・タンパク質間相互作用を大規模予測し、予測結果のいくつかは文献情報で確認することができました。
また米国食品医薬品局の有害報告システムの臨床データから薬物の副作用類似性を定義し、未知の薬物の標的分子やオフターゲット分子を網羅的に予測する方法を開発しました(Takarabe et al, Bioinformatics, 2012)。 これら一連の提案手法は薬効類似性や副作用類似性を利用した薬理ゲノミクスの新しい手法です。
ケモゲノミクスと薬理ゲノミクスの手法の予測結果は補完関係にあり、目的やデータに応じて使い分けていく必要があります(Yamanishi et al, Nucleic Acids Res, 2014)。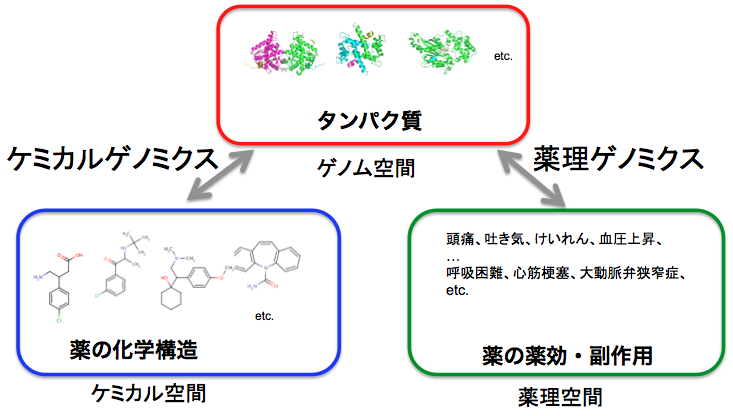
- 薬剤・化合物の毒性予測とメカニズム解析
-
薬剤の投与は、期待通りの治療効果をもたらす一方、望ましくない副作用を引き起こすこともあります。副作用は軽少なものから重篤なものまであり、医薬品開発において重要な懸案であるにも関わらず、その分子作用機構はほとんど未同定のままです。本研究では、薬剤-標的タンパク質の相互作用という分子レベルでの情報と、薬剤とその副作用という表現型レベルでの情報という、異なるレベルでの情報を関連づける方法を提案しました(Mizutani et al, Bioinformatics, 2012)。スパース正準相関解析という統計的手法を適用し、薬剤-標的タンパク質相互作用データと、薬剤-副作用データの間に相関のあるもののセットを抽出しました。下図は手法の概念図を示します。
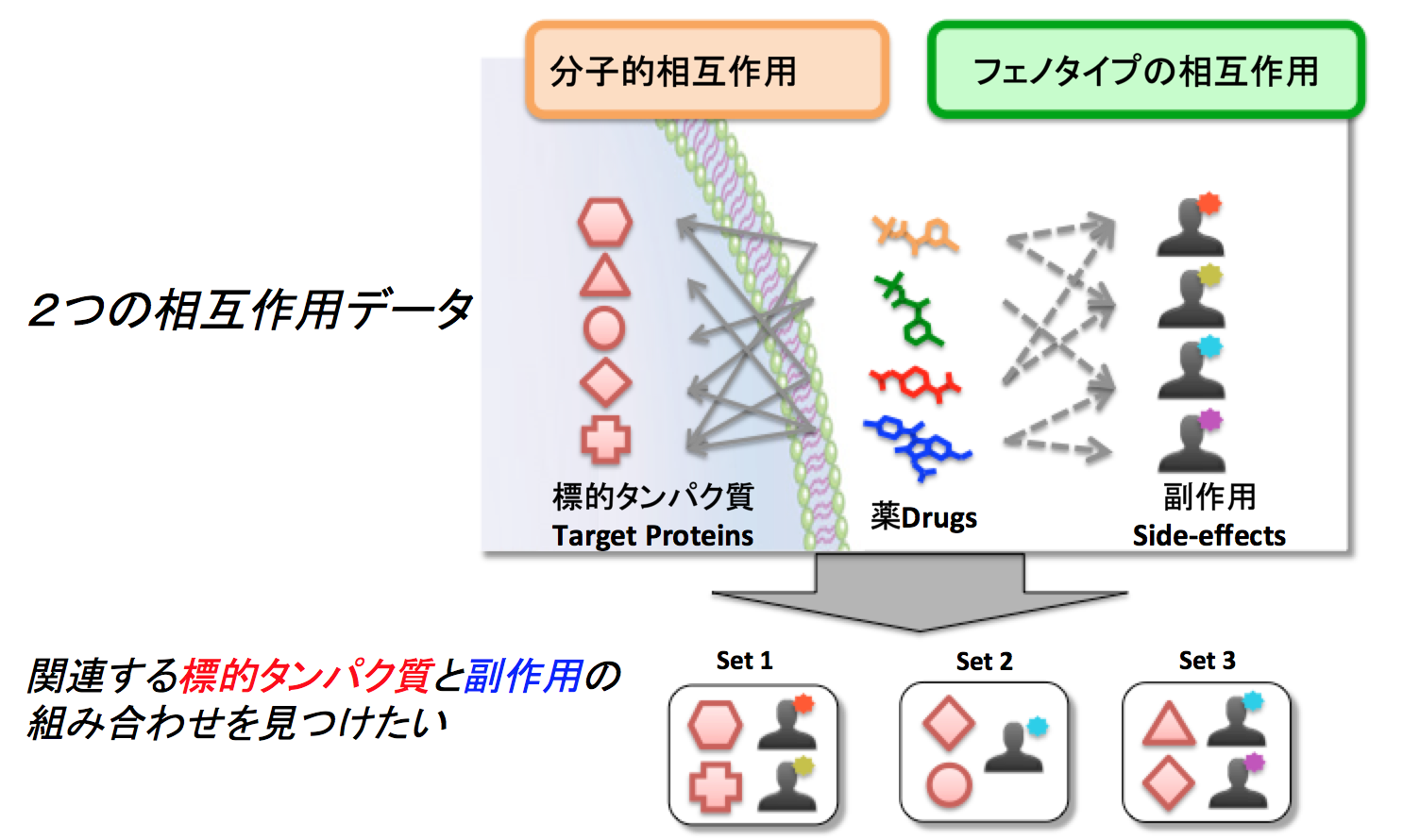
抽出された相関セットの生物学的妥当性は、Kyoto Encyclopedia of Genes and Genomes (KEGG) と Gene Ontology (GO) データベースを用いた Pathway Enrichment解析で評価しました。その結果、同じパスウェイで働くタンパク質は、分子機能が異なっていても同じ相関セットにクラスタリングされていることが分かりました。すなわち、ある相関セットにクラスタリングされたタンパク質の薬剤による機能制御は、同じパスウェイの活性、不活性化を通じて、同じような副作用を引き起こすのではないかと解釈することが出来ます。今回提案した手法は、副作用の分子作用機序への議論を可能にするとともに、薬剤候補化合物に対して標的タンパク質プロファイルから潜在的な副作用を予測するのに有用であると考えれます。また薬剤の化学構造情報と標的タンパク質情報を融合させた予測モデルを構築することで、副作用の予測精度が飛躍的に向上することが確認できました(Yamanishi et al, J Chem Inf Model, 2012)。
- 代謝パスウェイにおけるミッシング酵素遺伝子の同定
-
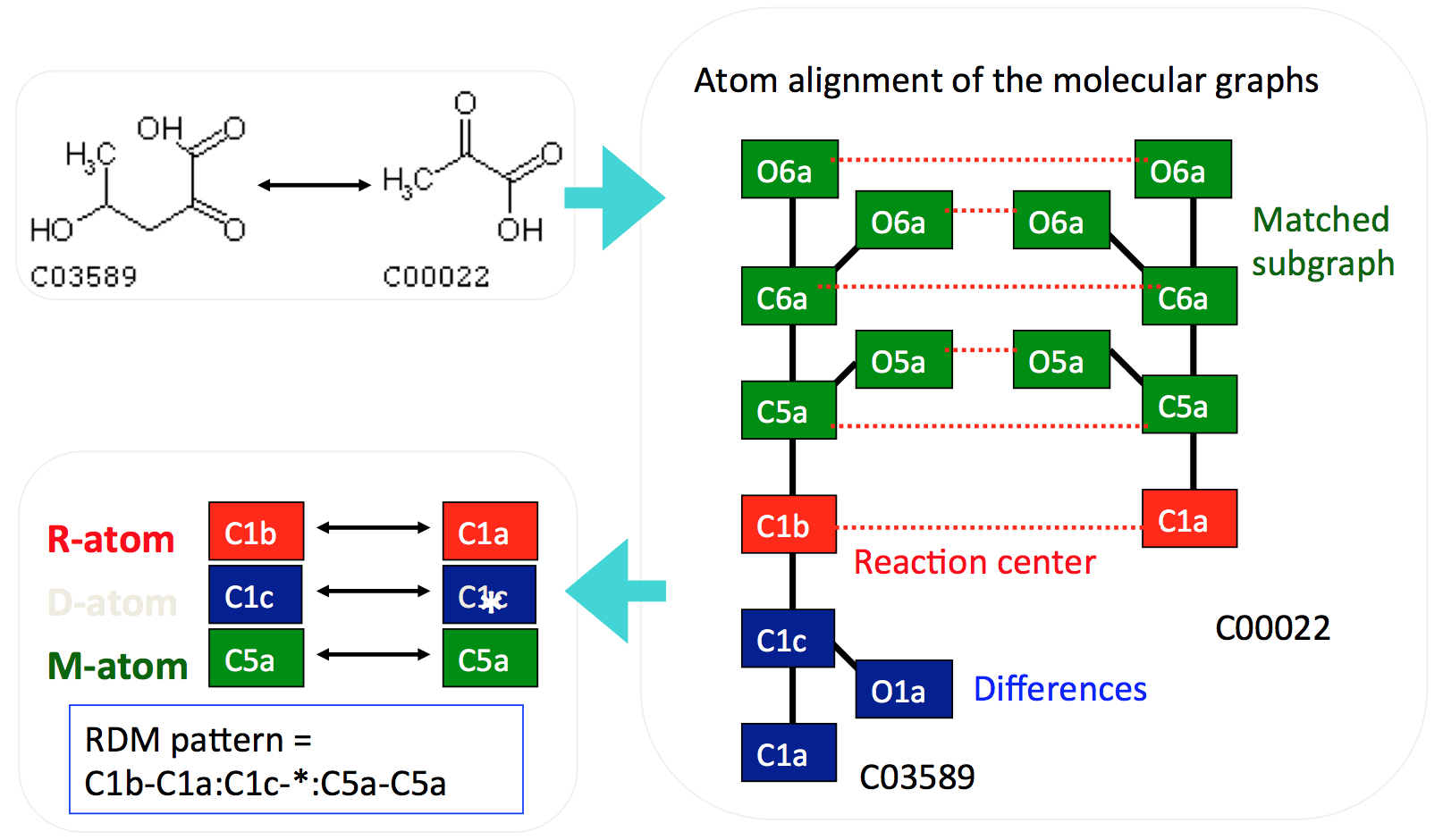
代謝パスウェイは、酵素と基質のネットワークと見なせます。本研究では、酵素遺伝子に関するゲノム関連データと基質に関するケミカル情報を組み合わせて、代謝パスウェイを予測する統計手法を開発しました。基質と生成物の間の部分構造変化に基づいて化学反応の分類パターンを提案し、任意の酵素反応に酵素番号を自動的に割り当てる統計手法を提案しました(Yamanishi et al, Bioinformatics, 2009)。下図は、基質と生成物の間の部分構造変化をRDMという化学反応分類パターンで表したものです(Kotera et al, J Am Chem Soc, 2004)。
また酵素番号の情報とゲノム関連データを組み合わせ、潜在的な化学反応経路を触媒する酵素遺伝子群を同定する方法を開発しました。提案手法の応用例として、緑膿菌のリジン分解系における反応の未知の酵素番号やミッシング酵素の候補遺伝子を系統プロファイルに基づく共進化類似性や遺伝子のゲノム上での並びに基づく遺伝子間距離から予測しました(Yamanishi et al, FEBS J, 2007)。下図は緑膿菌のリジン分解系のパスウェイで、L-lysineからGlutaryl-CoAまでの経路上にある箱が実際に予測したミッシング酵素群です。
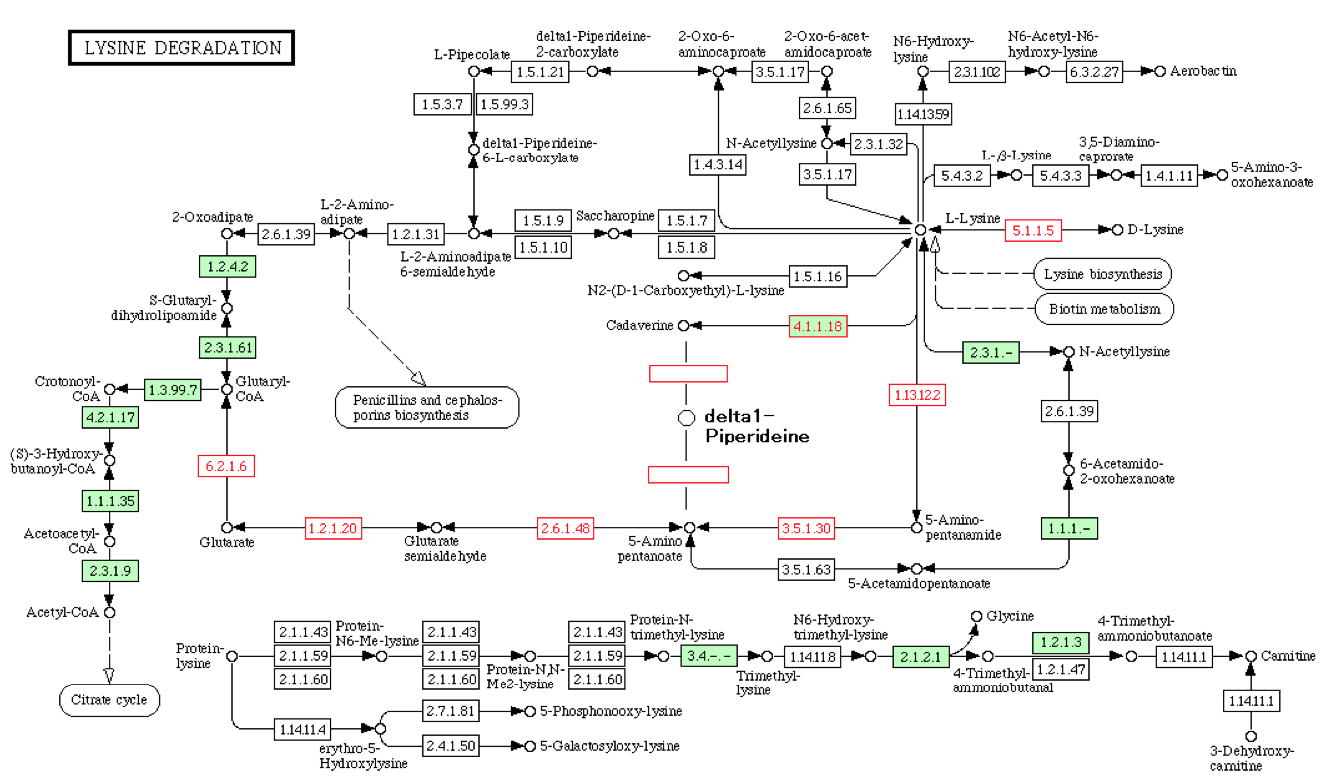
予測した酵素遺伝子に対し、実際に生化学的な実験で酵素活性を確認することで、新しい生物学的な発見に繋げることができました。これはドライとウェットの共同研究でもあります。これら一連の研究は、ケミカル情報とゲノム情報を繋げるケミカルゲノミクスの先駆的研究として世界的に位置づけられています。提案手法を実装したウェブサーバーを構築し、オンラインで利用可能な環境を提供しています。
- タンパク質間相互作用ネットワークの推定
-
生命システムは、遺伝子やタンパク質の機能的な相互作用ネットワークによって成り立っていますが、その大部分は未だに解明されていません。そのため様々なオミックスデータから未知の遺伝子間(またはタンパク質間)相互作用ネットワークを網羅的に予測することは挑戦的な課題です。遺伝子間またはタンパク質間の潜在的な機能的関係や代替パスなどの新しい生物学的発見につながることが期待されます。
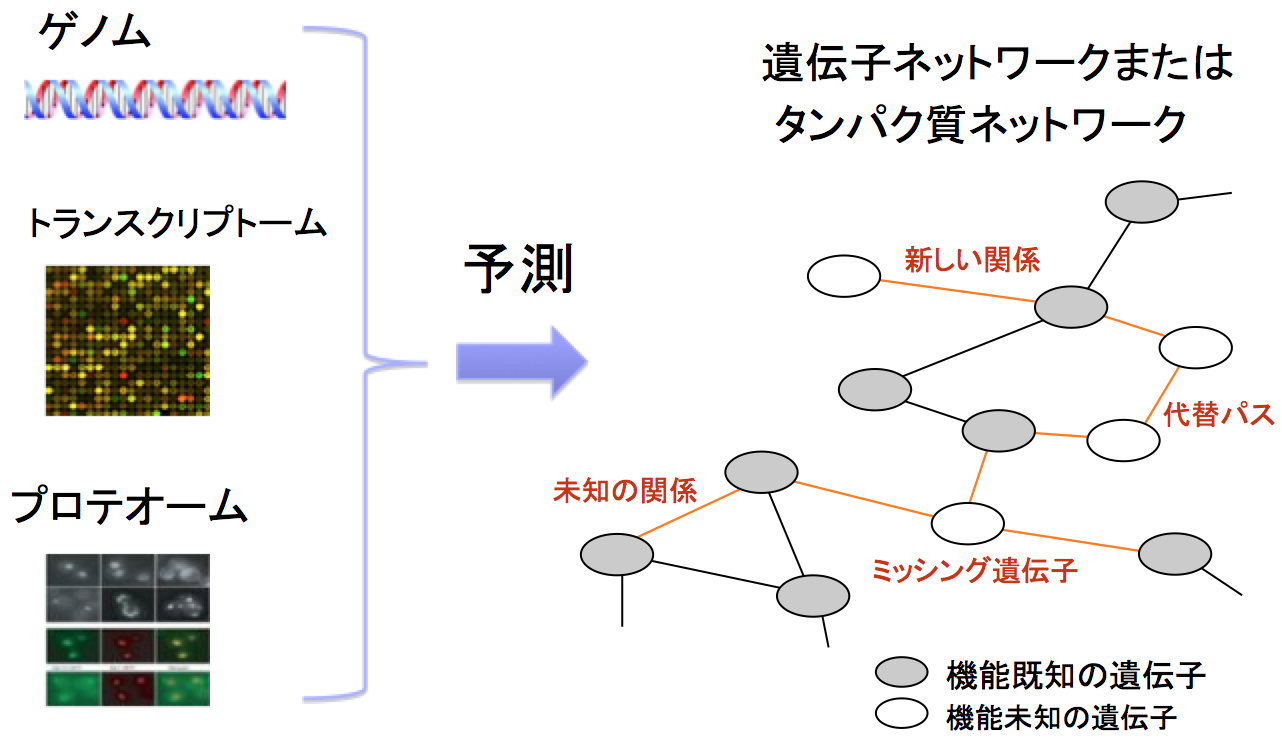
本研究では、遺伝子やタンパク質に関する様々な網羅的データから遺伝子間(またはタンパク質間)相互作用ネットワークを網羅的に予測する手法を開発しました(Yamanishi et al, Bioinformatics, 2004; Vert and Yamanishi, Adv Neural Inf Process Syst, 2005; Yamanishi et al, Bioinformatics, 2005)。データと相互作用ネットワークを表すグラフ構造の間の相関を統計モデルで学習し、それを用いて未知の相互作用を予測するという教師付き学習アルゴリズムである点が独創的です。また各データをカーネルとよばれる類似度行列に変換することで、異質なデータを融合してネットワークを予測する手順を提案しました。下図は提案したネットワーク予測法の概念図です。
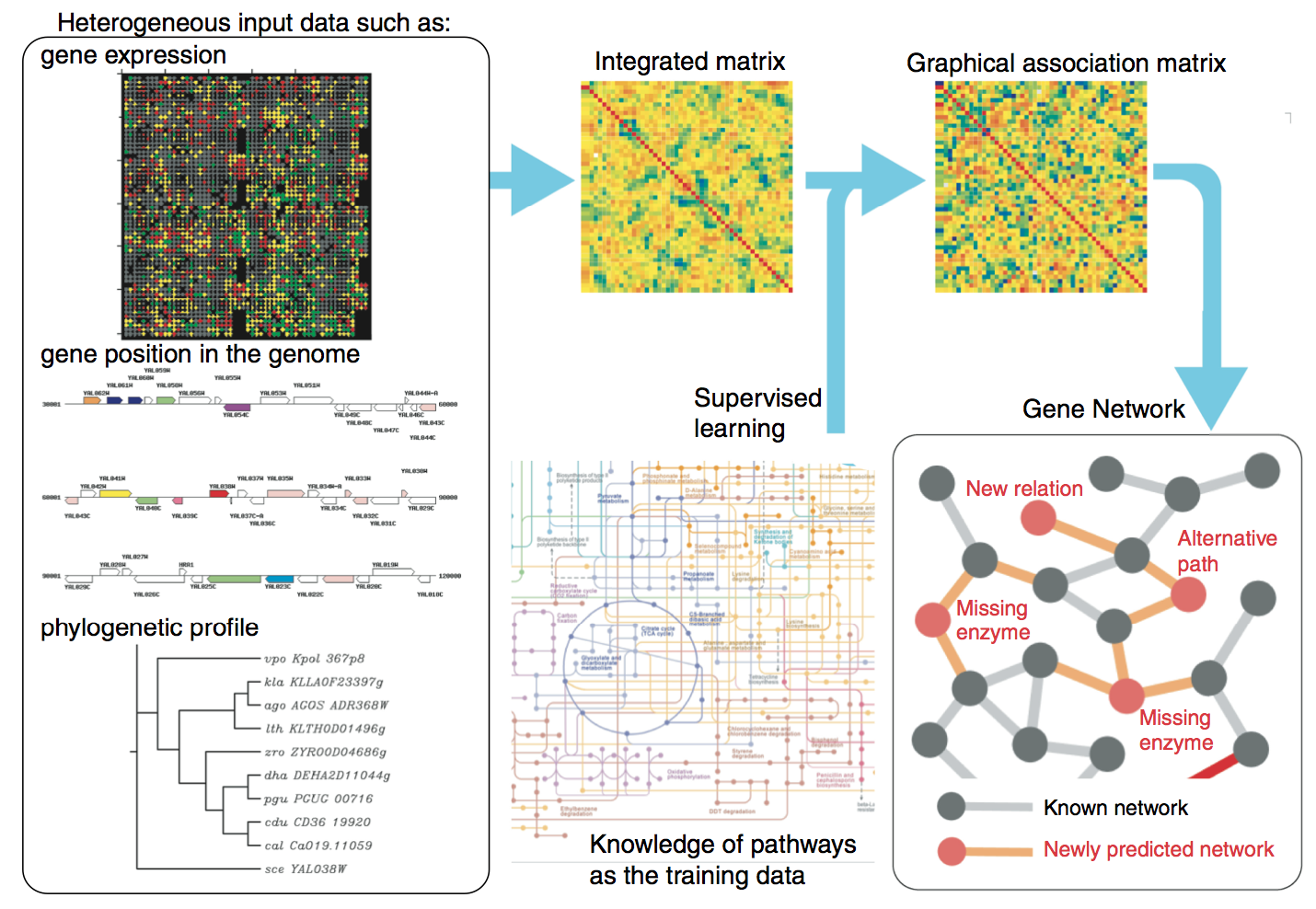
実際の適用例として、出芽酵母のタンパク質間相互作用ネットワークを、マイクロアレイ遺伝子発現情報、タンパク質の細胞内局在情報、系統プロファイルから予測し、クロスバリデーション実験でその有効性を示しています。これらの研究は、複数の異質な網羅的データの統合と教師付き学習に基づくタンパク質間相互作用ネットワーク予測の世界で初めて(当時)の研究でもあります。提案手法を実装したウェブサーバーを構築し、オンラインで利用可能な環境を提供しています(Kotera et al, Nucleic Acids Res, 2012)。
