Current research|現在の研究
※一部だけ紹介しています
※所属学生の卒業研究等の概要は、こちらのページ(学内限定)で紹介しています
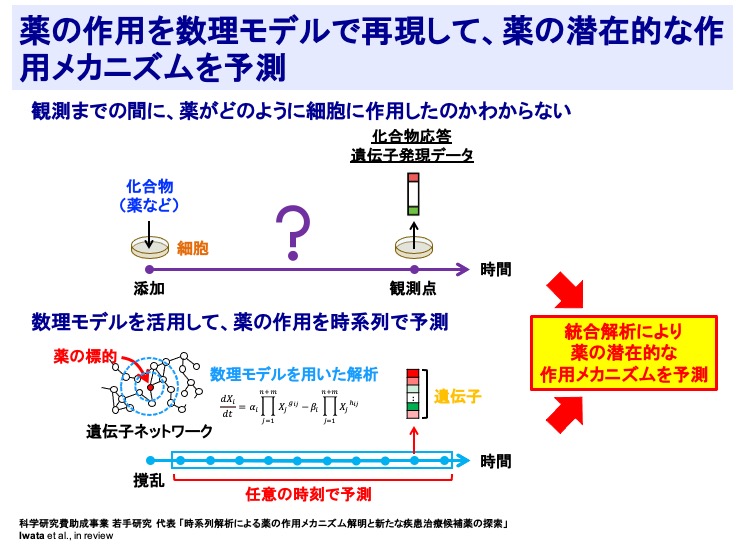
薬の作用を数理モデルで再現して、薬の潜在的な作用メカニズムを予測
化合物応答遺伝子発現データは、化合物を細胞に添加してから、一定時間経過後に観測されたデータですが、観測までの間に、薬がどのように細胞に作用したのかはわからないという問題がありました。そこで、数理モデルを活用して、薬の作用をコンピュータ上で再現することで、任意の時刻で、遺伝子発現データを予測する手法を開発しています。この手法により、観測値だけでは明らかにできなかった、薬の潜在的な作用メカニズムを予測することができるようになることが期待されます。
(Iwata et al., in revision)
Previous research|これまでの研究
※新しい研究成果から順に並んでいます
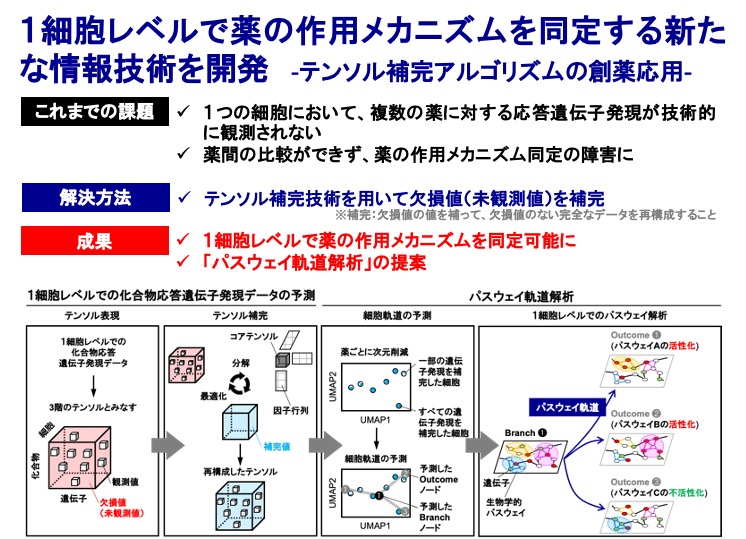
1細胞レベルで薬の作用メカニズムを同定する新たな情報技術を開発
これまでの創薬においては薬に対する細胞応答の不均一性は考慮されておらず、多様な細胞が混在した細胞集団に対して薬効を見出すことが一般的であり、1細胞レベルでの薬効のメカニズムを理解するのは困難でした。また、1細胞レベルでの遺伝子発現データには、測定技術の限界に起因する欠損値や未観測値が非常に多いという問題がありました。本研究では、1細胞レベルでの遺伝子発現データ解析において障害となっていた欠損値や未観測値を、高深度かつ高精度に補完するテンソル分解アルゴリズムを提案し、さらに1細胞レベルでの薬の作用メカニズムを生物学的パスウェイの視点から同定する点が特色です。開発手法を、膵臓内の膵島などで観測された1細胞レベルでの化合物応答遺伝子発現データに適用し、細胞タイプ特異的な薬の作用メカニズムの同定に成功しました。
(Iwata et al., Nat. Comput. Sci., 2022)
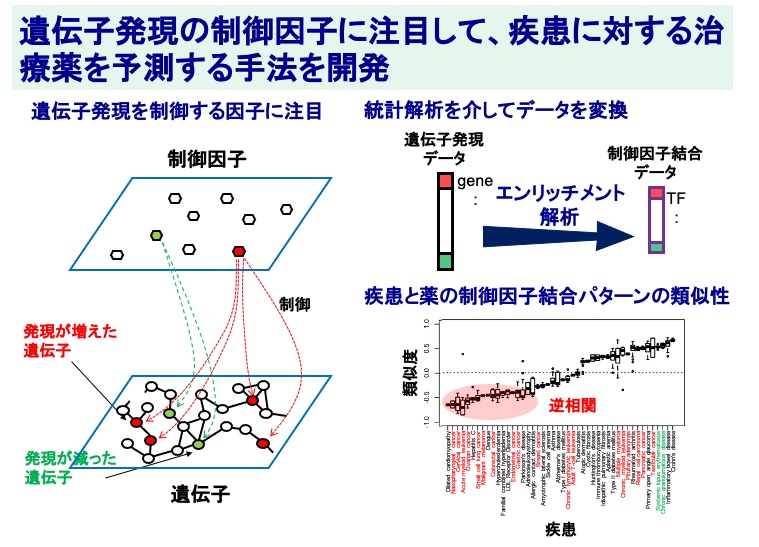
遺伝子発現の制御因子に注目して、疾患に対する治療薬を予測する手法を開発
疾患は、多くの遺伝子が協調的に相互作用する生体システムの破綻状態とみなすことができます。一方、疾患の治療薬は、破綻した遺伝子発現システムを正常な方向に制御するものです。したがって、最適な治療薬は疾患特異的な遺伝子発現パターンを打ち消す方向に働くと仮定して探索されてきました。また、これまで解析に用いてきた遺伝子発現データは、細胞内の遺伝子の情報しか見ていませんでしたが、本来は、それぞれの遺伝子の発現をなんらかの因子が制御しています。そこで、ビッグデータを用いた統計解析により、これまでの遺伝子発現データを、制御因子の情報からなる制御因子結合データに変換しました。この制御因子結合データを用いて、疾患と薬の制御因子結合パターンの類似性を調べることにより、さまざまながんに対して逆相関を用いた治療薬候補を予測できることが明らかとなりました。
(Iwata et al., npj Syst. Biol. Appl., 2022)
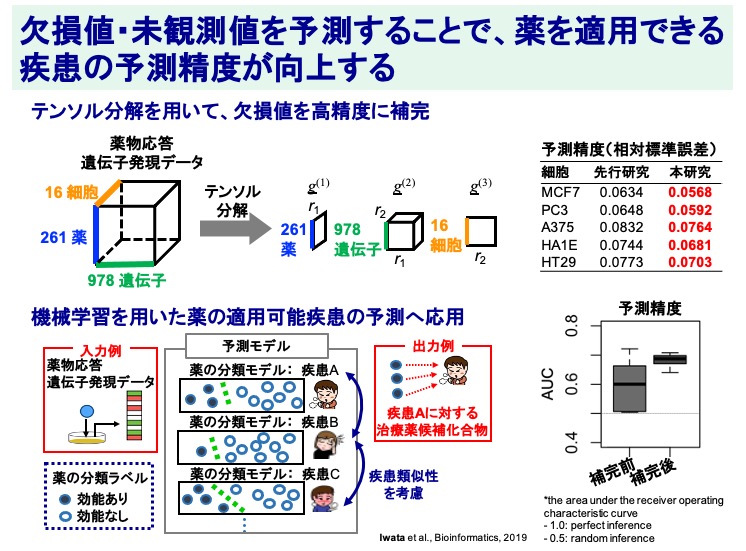
多様な細胞における化合物応答遺伝子発現を網羅的に予測する新技術を開発
化合物応答遺伝子発現データは創薬において非常に有用ですが、欠点として、すべての薬と細胞のペアに対してデータが得られないという点が挙げられます。我々は、薬、遺伝子、細胞からなるテンソルを構築して、その中で欠損値や未観測値を予測する手法を開発しました。行列データに対する予測である先行研究と比較して、テンソルデータに対する予測である本研究では、小さい誤差で正しい値を予測できることがわかりました。欠損値・未観測値を予測することで得られたデータは、創薬にも応用することができます。ここでは、機械学習を用いて、薬が適用可能な疾患を予測する問題を解いています。まず、それぞれの疾患に対して、その疾患に効能がある薬と効能がない薬を分けられるような予測モデルを立てて、薬の適用可能な疾患を予測しています。予測精度は、欠損値・未観測値を予測することで向上しており、創薬への有用性が明らかになりました。
(Iwata et al., Bioinformatics, 2019)
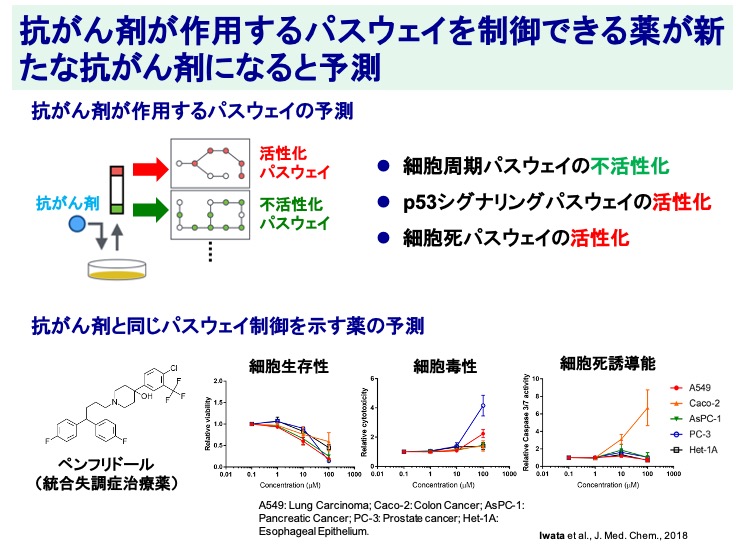
抗がん剤作用薬を自動的に予測する情報技術を開発
既存の抗がん剤によって、発現が増えた遺伝子群と発現が減った遺伝子群を解析し、抗がん剤によって活性化されるパスウェイと不活性化されるパスウェイを同定しました。その結果、ここに示す、細胞周期パスウェイの不活性化、p53シグナリングパスウェイの活性化、細胞死パスウェイの活性化が、多くの抗がん剤で制御されていることを同定しました。これらの制御は、従来の抗がん剤の作用メカニズムにも合致するものです。そこでわれわれは、逆に、これらのパスウェイ制御を示す薬は、新たな抗がん剤として使用できるのではないかと予測しました。上位に予測された薬のうち、統合失調症治療薬であるペンフリドールは、細胞生存性や細胞毒性、細胞死誘導能の観点で、実験的にも有望な抗がん剤候補として同定することができました。
(Iwata et al., J. Med. Chem., 2018)
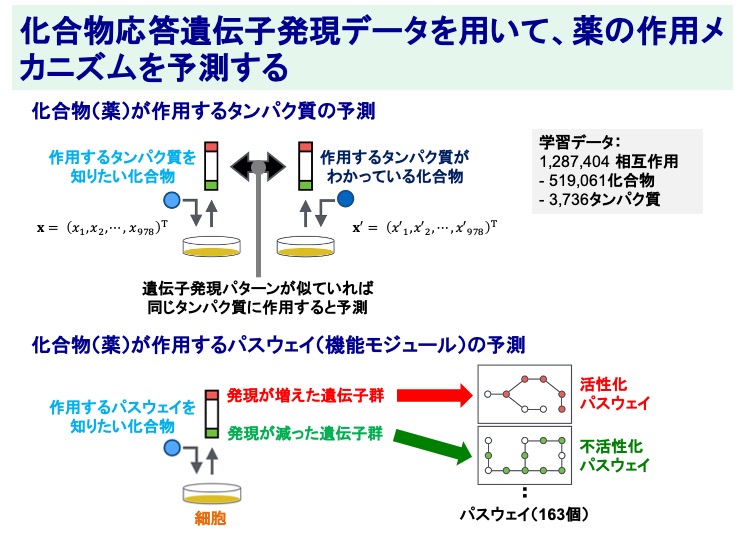
化合物応答遺伝子発現データを用いて、薬の作用メカニズムを予測する
創薬における課題として、研究開発費用が年々増加している一方で、承認される新薬の数が減少傾向にあるという点が挙げられます。解決策として考えられるのは、1から新薬を創るのではなく、すでに承認されている薬の中から、別の疾患にも使える薬を探すというアプローチです。このためには、承認薬を含むさまざまな生物活性化合物の細胞への影響を、遺伝子レベルで観測して、得られたデータ(化合物応答遺伝子発現データ)を解析することが有用です。このデータを用いて、薬の作用メカニズムを予測することができます。薬は、細胞内のタンパク質に作用して、治療効果を発揮するため、そのタンパク質を明らかにすることは重要です。薬が作用するタンパク質を予測したい場合には、「作用するタンパク質を知りたい化合物」と「作用するタンパク質がわかっている化合物」をそれぞれ細胞に振りかけたときに得られた遺伝子発現パターンを比較します。もし、遺伝子発現パターンが似ていれば、どちらの化合物も同じタンパク質に作用するのではないかと予測できます。また、タンパク質1つ1つではなく、複数のタンパク質からなる機能モジュールであるパスウェイを予測することも有用です。この場合には、「作用するタンパク質を知りたい化合物」によって、発現が増えた遺伝子群と発現が減った遺伝子群から、統計解析により、どのようなパスウェイを活性化して、不活性化するのか予測することができます。
(Iwata et al., Sci. Rep., 2017)
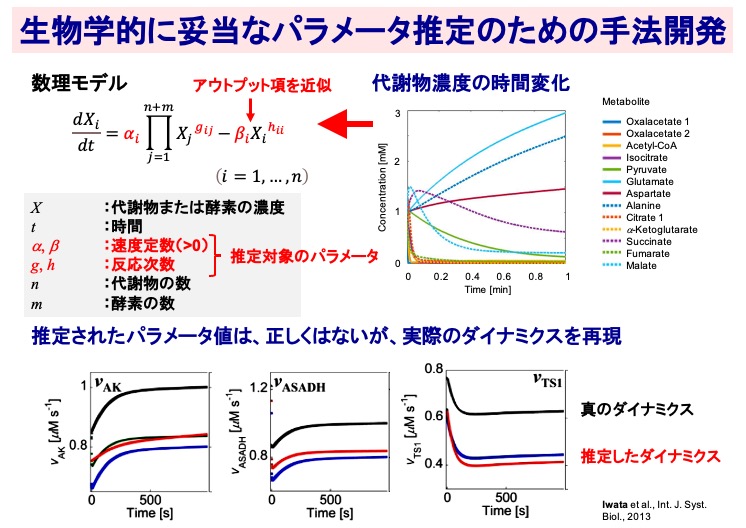
生物学的に妥当なパラメータ推定のための手法開発
生物がもつシステムには曖昧さが存在し、測定される代謝物の濃度にも誤差が含まれるため、より生物学的に妥当なパラメータを推定する手法の開発が求められました。そこで、数理モデルのアウトプット項を近似して、より頑健かつ実用的な数理モデル推定法を開発しました。この手法により得られるパラメータ値は、真の値とは一致しませんが、生物学的な指標で時系列のダイナミクスを評価すると、実際のダイナミクスを再現できることがわかりました。ここでは、3つの指標について、黒で示す「真のダイナミクス」と、赤や青で示す「推定したダイナミクス」が同じような挙動を示していることがわかります。これらのことから、パラメータ推定においては、必ずしも正しい値を予測する必要がない可能性が示唆されました。
(Iwata et al., Int. J. Syst. Biol., 2013)
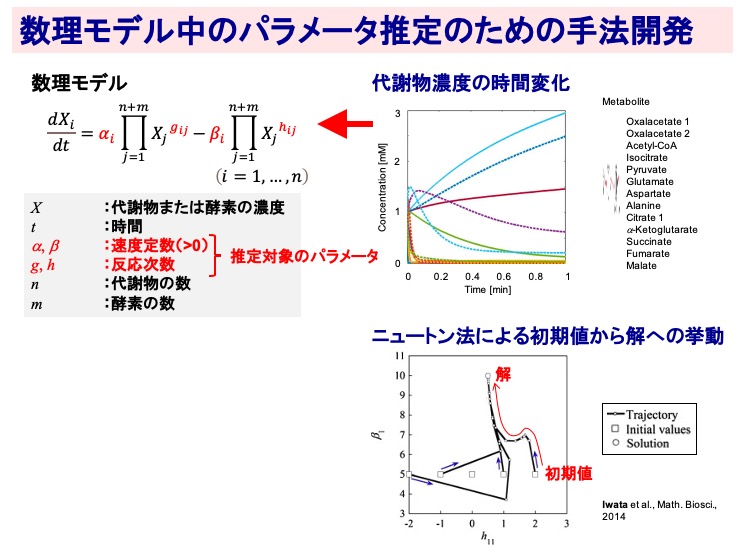
数理モデル中のパラメータ推定のための手法開発
代謝物濃度の時間変化のデータが与えられたときに、そのデータを用いて、数理モデルに含まれるパラメータを推定するための手法を開発しました。これは、一般のパラメータフィッティングの問題になりますが、この形状の数理モデルに対して最適な方法が確立されていないという状況があり、私たちは、解を探索する手法として有名なニュートン法を採用して、与えた初期値から解へと迅速に収束できる手法を開発しました。
(Iwata et al., Math. Biosci., 2014)
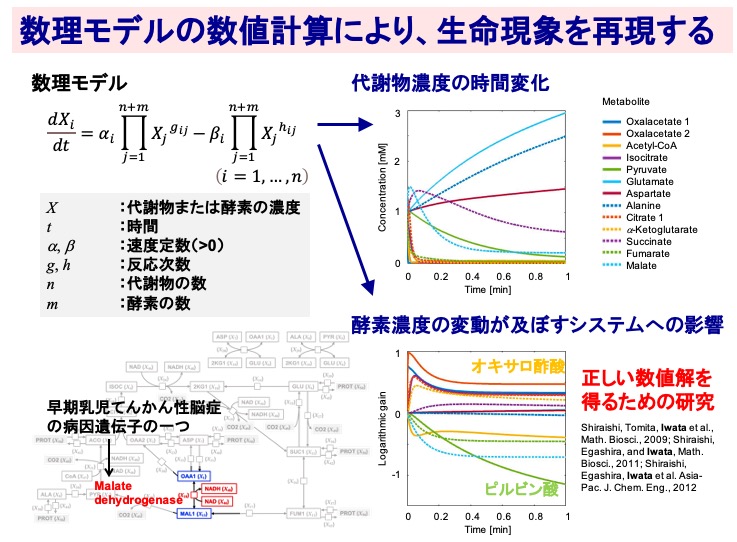
数理モデルの数値計算により、生命現象を再現する
生物を構成する1つ1つの細胞は、非常に複雑な分子間制御ネットワークを持っており、ネットワークの特性を理解するために、数理モデルを構築して解析することが有用です。例えば、ある代謝物の濃度の時間変化を、その代謝物を生成する代謝物や酵素からなるインプット項、その代謝物を分解する酵素などからなるアウトプット項で表現することができます。一旦数理モデルが構築されれば、数値計算を行うことにより、それぞれの代謝物の濃度がどのように時間変化するのか、ある酵素の濃度を変えたときに代謝反応システムがどのような影響を受けるのか、コンピュータ上でシミュレーションすることができます。この例では、早期乳児てんかん性脳症の病因遺伝子とされる酵素を変動させたときに、それぞれの代謝物がどのような影響を受けるのかを計算しています。
(Shiraishi, Tomita, Iwata et al., Math. Biosci., 2009; Shiraishi, Egashira, and Iwata, Math. Biosci., 2011; Shiraishi, Egashira, Iwata et al. Asia-Pac. J. Chem. Eng., 2012)